A Comprehensive Guide to qRT-PCR Validation of Vitellogenin Knockdown: From Foundational Principles to Clinical Applications
This article provides a comprehensive framework for researchers validating vitellogenin (Vg) gene knockdown using quantitative real-time PCR (qRT-PCR).
A Comprehensive Guide to qRT-PCR Validation of Vitellogenin Knockdown: From Foundational Principles to Clinical Applications
Abstract
This article provides a comprehensive framework for researchers validating vitellogenin (Vg) gene knockdown using quantitative real-time PCR (qRT-PCR). Vitellogenin, a critical protein in reproduction and other physiological processes, is a key target in functional genetics and pest control research. We cover the foundational biology of Vg, detailed methodological protocols for RNAi and qRT-PCR analysis, advanced troubleshooting for assay optimization, and rigorous validation strategies. Adhering to MIQE guidelines and the fit-for-purpose concept, this guide is essential for ensuring accurate, reproducible, and clinically relevant data in studies ranging from insect physiology to the development of novel biocontrol agents.
Understanding Vitellogenin: Biology, Function, and Rationale for Knockdown
The Multifunctional Role of Vitellogenin in Reproduction, Immunity, and Longevity
Vitellogenin (Vg) is an evolutionarily conserved glycolipoprotein found in nearly all oviparous species, traditionally recognized as the primary precursor to egg yolk proteins [1]. However, contemporary research has unveiled a remarkable expansion of its functional portfolio, positioning Vg as a multifunctional protein with significant roles in immunity, longevity, and social behavior regulation, far beyond its canonical reproductive purpose [2] [3] [4]. This functional pleiotropy is particularly pronounced in social insects like the honey bee (Apis mellifera), where Vg has been co-opted into regulatory networks governing complex social traits [2] [5]. The investigation of Vg's diverse roles relies heavily on robust gene manipulation and validation techniques, with qRT-PCR serving as a cornerstone for confirming gene knockdown efficacy in functional studies. This article examines the comparative functions of Vg across insect species, with a specific focus on experimental approaches for validating Vg manipulations and their consequent phenotypic effects.
Comparative Functional Analysis of Vitellogenin Across Insect Species
Table 1: Comparative Analysis of Vitellogenin Functions Across Insect Species
| Species | Reproductive Role | Immunological Function | Impact on Longevity | Behavioral Influence | Key Experimental Evidence |
|---|---|---|---|---|---|
| Honey bee (Apis mellifera) | Nutrient source for brood food [5] | Antioxidant activity; possible pathogen recognition [3] | Increased lifespan [2] [6] | Regulates foraging onset & specialization; influences swarming [2] [5] | RNAi knockdown; qRT-PCR validation [2] [6] |
| Red palm weevil (Rhynchophorus ferrugineus) | Major yolk precursor; essential for oogenesis [7] | Not specifically studied | Not reported | Not reported | RNAi knockdown leads to atrophied ovaries [7] |
| Kissing bug (Rhodnius prolixus) | Essential for embryo development [4] | Not the focus of study | Increased lifespan after silencing [4] | Not reported | RNAi of Vg1 & Vg2 isoforms [4] |
| Diamondback moth (Plutella xylostella) | Vg transport via VgR crucial for oocyte development [8] | Not reported | Not reported | Not reported | CRISPR/Cas9-mediated VgR knockout [8] |
Table 2: Quantitative Effects of Vitellogenin Gene Manipulation on Phenotypic Traits
| Species | Intervention | Effect on Reproduction | Effect on Lifespan | Effect on Behavior | Validation Method |
|---|---|---|---|---|---|
| Honey bee | Vg RNAi | Not primary focus | Reduced lifespan [2] | Earlier foraging onset; nectar preference [2] | qRT-PCR [6] |
| Red palm weevil | Vg RNAi | 95-99% Vg suppression; atrophied ovaries; no viable eggs [7] | Not reported | Not reported | qRT-PCR, SDS-PAGE [7] |
| Kissing bug | Vg1/Vg2 RNAi | Yolk-depleted eggs; most eggs inviable [4] | Increased in both males and females [4] | Not reported | qRT-PCR, phenotypic observation [4] |
| Diamondback moth | VgR knockout | Smaller, whiter eggs; lower hatch rate [8] | Not reported | Not reported | CRISPR/Cas9, sequencing [8] |
Experimental Protocols for Vitellogenin Functional Studies
RNA Interference (RNAi) and qRT-PCR Validation Protocol
RNAi-mediated gene silencing has emerged as a powerful tool for investigating Vg function. The following protocol outlines the key steps for Vg knockdown and validation, as demonstrated in honey bee studies [2] [6] and Red palm weevil research [7]:
dsRNA Design and Synthesis: Primers are designed from the target species' Vg cDNA sequence (e.g., GenBank AJ517411 for A. mellifera) and fused with T7 promoter sequences. The Green Fluorescent Protein (GFP) gene is commonly used as a dsRNA control to account for non-specific effects of injection and dsRNA presence [2] [6]. PCR amplification using the Vg template generates a product, which is purified and used for dsRNA synthesis with systems like the RiboMax T7 system [6]. The resulting dsRNA is purified, resuspended in nuclease-free water, and quality is checked via agarose gel electrophoresis [6] [7].
Delivery of dsRNA: Newly emerged adult worker bees are briefly cold-anesthetized and injected with 2 µL of dsRNA solution (e.g., 5 µg/µL) dorsally between the fifth and sixth abdominal segments using a micro-syringe with a G30 needle [6]. Control groups receive GFP dsRNA, and a non-injected group may serve as an additional reference [2] [6].
Validation of Knockdown via qRT-PCR: Total RNA is extracted from target tissues (e.g., fat body or abdomen) using commercial kits (e.g., TRIzol Reagent or Maxwell RSC SimplyRNA Tissue Kit) [5] [7]. RNA quality and concentration are assessed spectrophotometrically. cDNA is synthesized using reverse transcriptase (e.g., HiscriptTM Reverse Transcriptase) [8]. qPCR is performed using a real-time PCR system (e.g., Bio-Rad CFX Connect) with gene-specific primers for Vg and reference genes (e.g., β-actin and NDUFA8 for honey bees) [5]. The reaction mixture typically includes SYBR/FAM dye. The thermal cycling profile includes an initial denaturation (e.g., 95°C for 3 min), followed by 40 cycles of denaturation (e.g., 95°C for 5-30 s), annealing (temperature specific to primers, e.g., 57.5°C for honey bee Vg), and extension (e.g., 72°C for 10 s) [5]. Relative gene expression is calculated using the ΔΔCt method, normalizing to reference genes and comparing to control groups [5].
CRISPR/Cas9-Mediated Gene Editing
For the vitellogenin receptor (VgR), CRISPR/Cas9 has been successfully applied in the Diamondback moth [8]:
Target Selection and gRNA Design: A specific target site within the VgR gene exon is selected. Single-guide RNAs (sgRNAs) are designed and transcribed in vitro.
Microinjection: A mixture of Cas9 mRNA and sgRNA is microinjected into freshly laid eggs.
Mutant Screening: Surviving G0 adults are backcrossed, and their progeny (G1) are screened for mutations. Genomic DNA is extracted from individual insects, the target region is PCR-amplified, and products are sequenced to identify indel mutations, such as a 5-bp deletion [8].
Validation of Functional Impact: The phenotypic consequences are assessed by examining VgR protein expression in ovaries via immunohistochemistry or Western blot, observing ovarian development, egg morphology, and egg hatch rates [8].
Figure 1: Vitellogenin Regulatory Network in Honey Bees. This diagram illustrates the central role of vitellogenin in a feedback loop with juvenile hormone, and its pleiotropic effects on behavior, lifespan, and immunity [2] [6].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Vitellogenin Functional Studies
| Reagent / Solution | Function in Experiment | Specific Examples / Notes |
|---|---|---|
| Vg-specific dsRNA | Triggers sequence-specific degradation of Vg mRNA | Designed from species-specific Vg cDNA (e.g., A. mellifera AP4a5 clone) [2] [6] |
| Control dsRNA (e.g., GFP) | Control for injection procedure and non-specific immune response | GFP dsRNA does not share close homology with insect genes [6] |
| TRIzol Reagent | RNA isolation from tissues (fat body, ovary) | Maintain RNA integrity for accurate qRT-PCR results [7] [8] |
| Reverse Transcriptase | Synthesis of cDNA from RNA template | Essential step for qRT-PCR and gene expression analysis [8] |
| qPCR Primers (Vg & Reference Genes) | Amplification of specific transcripts for quantification | Reference genes (e.g., β-actin, NDUFA8) are crucial for normalization [5] |
| SYBR Green/FAM Dye | Fluorescent detection of amplified DNA during qPCR | Allows real-time monitoring of PCR product accumulation [5] |
| Cas9 Protein/mRNA & sgRNA | CRISPR/Cas9-mediated gene knockout of Vg or VgR | Used for creating stable genetic mutants [8] |
| Microinjection Apparatus | Delivery of dsRNA or CRISPR components into insects | Requires precision needles (e.g., G30) and micromanipulators [6] [8] |
Figure 2: Vitellogenin Functional Study Workflow. This flowchart outlines the key steps in a typical RNAi-based functional study, from dsRNA preparation to phenotypic validation [2] [6] [7].
Vitellogenin exemplifies the evolutionary adaptability of a fundamental reproductive protein to acquire diverse physiological roles. The experimental data consistently demonstrate that Vg is indispensable for reproduction across all insect species studied. However, its co-option as a central regulator of social behavior, lifespan, and immunity appears most developed in the honey bee, a model for complex social organization [2] [5]. The efficacy of RNAi and CRISPR/Cas9 in disrupting Vg signaling and causing profound phenotypic changes, particularly reproductive failure, also highlights the potential of Vg and its pathway components as promising targets for developing novel, species-specific pest control strategies [7] [8]. Future research leveraging these sophisticated genetic tools will continue to unravel the intricate mechanisms by which this multifunctional protein coordinates complex life-history traits.
Vitellogenin (Vg), a precursor of yolk proteins, is a critical component in the reproductive processes of oviparous animals, including insects, fish, and other invertebrates. Its role in vitellogenesis—the process of yolk formation—makes it an essential factor for embryonic development and reproductive success. In recent years, Vg has emerged as a promising genetic target for pest control strategies and functional genomics research. The suppression of Vg gene activity disrupts egg development and reduces fertility, offering a species-specific approach for managing agriculturally and medically significant pests. This guide provides a comparative analysis of Vg-targeting methodologies, synthesizes quantitative experimental data, and outlines essential protocols and reagents, framed within the context of qRT-PCR validation for Vg knockdown research.
Vitellogenin Function and Conservation
Vitellogenin is a conserved lipoprotein that serves as the primary precursor to vitellin (Vn), the major yolk protein stored in oocytes. It is synthesized in the fat body (in insects) or liver (in fish), released into the hemolymph or bloodstream, and transported to developing oocytes where it is internalized via the vitellogenin receptor (VgR) [9] [8]. The internalized Vg is then processed into Vn, which provides crucial nutrients for embryonic growth.
Molecular analysis reveals that Vg proteins share conserved structural domains across species. These typically include:
- A lipoprotein N-terminal domain (LPD_N)
- A domain of unknown function (DUF1943)
- A von Willebrand factor type D domain (vWD) [10] [11]
The molecular characterization of Vg is a critical first step in designing targeted control strategies, as these conserved regions can be exploited for the development of specific inhibitory agents.
Methodologies for Targeting Vitellogenin
RNA Interference (RNAi)
RNAi is a widely used technique for silencing gene expression by introducing sequence-specific double-stranded RNA (dsRNA), which leads to the degradation of complementary mRNA.
Protocol: RNAi-Mediated Vg Knockdown
- Target Gene Identification: Clone and sequence the full-length Vg transcript from the target species (e.g., Rhynchophorus ferrugineus yielded a 5504 bp transcript) [7].
- dsRNA Synthesis: Design primers with T7 promoter sequences to amplify a unique, species-specific fragment of the Vg gene (e.g., 504 bp fragment for R. ferrugineus). Use this PCR product as a template for in vitro transcription with a system like the Promega RiboMax T7 to generate dsRNA [7].
- dsRNA Delivery:
- qRT-PCR Validation:
- RNA Extraction: After a set period (e.g., 15-25 days post-injection), extract total RNA from the fat body or relevant tissues using TRIzol Reagent.
- cDNA Synthesis: Synthesize cDNA using a reverse transcriptase kit (e.g., SensiFAST cDNA kit).
- qPCR Amplification: Perform qRT-PCR with a SYBR Green kit (e.g., SensiFAST SYBR Green Kit) on a real-time cycler (e.g., BioRad CFX96). Use stable reference genes (e.g., pmp-3 for C. elegans, tubulin for insects) for normalization [12] [7].
- Phenotypic Assessment: Monitor changes in fecundity, egg hatchability, and ovarian development.
CRISPR/Cas9 Gene Editing
CRISPR/Cas9 enables permanent genomic disruption of the Vg gene, allowing for the analysis of its heritable loss-of-function effects.
Protocol: CRISPR/Cas9-Mediated Vg Knockout
- sgRNA Design: Identify a 20-nucleotide target sequence adjacent to a Protospacer Adjacent Motif (PAM: NGG) in an exon of the Vg gene. This sequence should be unique to the target gene to minimize off-target effects [8] [13].
- sgRNA and Cas9 Preparation: Synthesize sgRNA in vitro and purify it. Use commercially available Cas9 nuclease protein or mRNA.
- Microinjection: Co-inject sgRNA and Cas9 into newly laid embryos (zygotes) using a microinjection system [8] [13].
- Mutant Screening:
- Raise the injected (F0) generation, which will be mosaic. Outcross F0 adults to wild-type individuals.
- Extract genomic DNA from F1 offspring and screen for mutations at the target site using PCR and sequencing. Identify heterozygous carriers.
- Cross heterozygous (F1) individuals to generate homozygous (F2) mutants for analysis [13].
- Validation:
Comparative Efficacy of Vg-Targeting Strategies
The following tables summarize experimental data from various studies, highlighting the efficacy of different Vg-targeting strategies across species.
Table 1: Efficacy of RNAi-Mediated Vitellogenin Knockdown in Pest Control
| Pest Species | Target Gene | Delivery Method | Knockdown Efficiency | Biological Impact | Reference |
|---|---|---|---|---|---|
| Red Palm Weevil(Rhynchophorus ferrugineus) | RfVg | Injection (dsRNA) | 95-99% (qRT-PCR) | Failed oogenesis, atrophied ovaries, no egg hatch | [7] |
| Honey Bee(Apis mellifera) | Vg | Injection (dsRNA) | Significant reduction (qRT-PCR) | Increased gustatory responsiveness; accelerated behavioral maturation | [6] |
| Ant(Temnothorax longispinosus) | Vg-like A | Injection (dsiRNA) | Significant reduction (RNAseq/qPCR) | Reduced brood care, increased nestmate care | [14] |
| C. elegans(Model Organism) | vit-2 | Feeding (RNAi) | Significant upregulation in pry-1 mutant rescued (qPCR) | Rescued lipid levels & lifespan defect | [12] |
Table 2: Efficacy of CRISPR/Cas9-Mediated Vitellogenin Gene Disruption
| Species | Target Gene | Mutation Type | Molecular & Phenotypic Consequences | Biological Impact | Reference |
|---|---|---|---|---|---|
| Zebrafish(Danio rerio) | vtg2 | 2811 bp deletion (Frameshift) | 5x reduced Vtg2 in liver; 3.8x reduced in embryos (LC-MS/MS) | 29% survival at 24 hpf; yolk leakage, morphological abnormalities | [13] |
| Diamondback Moth(Plutella xylostella) | VgR | 5 bp deletion (Frameshift) | VgR protein deficiency in ovaries & eggs | Smaller, whiter eggs; lower egg hatching rate | [8] |
| Eggplant Shoot & Fruit Borer(Leucinodes orbonalis) | LoVg | Not specified | Not specified | No effect on egg laying; severe impact on egg hatchability | [11] |
The Scientist's Toolkit: Essential Research Reagents
Successful investigation of vitellogenin requires a standardized set of reagents and methodologies. The following table details key solutions for Vg-focused research.
Table 3: Essential Research Reagent Solutions for Vitellogenin Studies
| Reagent / Kit | Primary Function | Example Use Case | Citation |
|---|---|---|---|
| TRIzol Reagent | Total RNA isolation from tissues (fat body, liver, ovary) | RNA extraction for downstream qRT-PCR validation | [12] [8] |
| SensiFAST cDNA Synthesis Kit | High-efficiency synthesis of first-strand cDNA from RNA templates | Preparation of cDNA for qPCR amplification | [12] |
| SensiFAST SYBR Green Kit | Sensitive detection and quantification of DNA amplification in qPCR | qRT-PCR analysis of Vg transcript levels | [12] |
| RiboMax T7 System | Large-scale in vitro synthesis of dsRNA for RNAi experiments | Production of dsRNA targeting Vg mRNA | [7] |
| Hamilton Micro-Syringe | Precise microinjection of dsRNA or CRISPR components into insects/embryos | Delivery of genetic material for RNAi and CRISPR protocols | [6] [7] |
| pJET1.2 Vector | High-efficiency cloning of PCR products for sequencing | Verification of Vg gene sequences and CRISPR-induced mutations | [8] |
Vitellogenin Signaling and Experimental Workflow
The diagram below illustrates the fundamental role of Vg in reproduction and the primary mechanisms for its genetic disruption.
Diagram 1: Vitellogenin pathway and genetic disruption mechanisms. The normal pathway (green) shows Vg synthesis, transport, and utilization. Genetic disruption techniques (red) target Vg at the gene (CRISPR) or mRNA (RNAi) level to inhibit reproduction.
The following diagram outlines a generalized experimental workflow for developing and validating a Vg-targeting strategy.
Diagram 2: Experimental workflow for Vg-targeted functional genomics and pest control. This pipeline guides researchers from initial gene discovery to final validation of a Vg-targeting intervention.
Vitellogenin represents a genetically tractable and highly effective target for regulating reproduction in pest species. As the comparative data shows, both RNAi and CRISPR/Cas9 methodologies can achieve profound suppression of Vg function, leading to significant reductions in fertility and population growth. The choice of technique depends on the target organism, desired persistence of the effect, and available resources. RNAi offers a reversible, non-heritable suppression suitable for bait-based control strategies, while CRISPR/Cas9 provides a permanent, heritable solution with potential for gene drive applications. The consistent success of these approaches across diverse species underscores the conserved essentiality of Vg in reproduction. Future work will focus on optimizing delivery mechanisms, enhancing species specificity, and navigating the regulatory landscape for the field application of these powerful genetic technologies.
Vitellogenin (Vg), a glycolipoprotein traditionally known as a yolk precursor, plays a surprisingly diverse set of roles in animal physiology. While its function in reproduction is well-documented across oviparous species, recent research utilizing targeted gene knockdown approaches, particularly RNA interference (RNAi), has revealed its critical involvement in processes ranging from thermal stress protection to complex social behaviors. This guide systematically compares the biological consequences of Vg knockdown across multiple experimental models, providing researchers with a consolidated overview of phenotypic outcomes and methodological approaches essential for designing robust functional studies, particularly those employing qRT-PCR for validation.
Comparative Phenotypic Outcomes of Vg Knockdown
Table 1: Documented Effects of Vg/Vg-like Gene Knockdown Across Species
| Species | Key Biological Process Impacted | Knockdown Method | Major Phenotypic Outcome | Experimental Validation |
|---|---|---|---|---|
| Mud Crab (Scylla paramamosain) [15] | Oocyte heat stress protection | Natural mutation (Enhancer deletion) | Impaired vitellogenic oocyte formation at high temperatures (>30°C); Failure of Vtg uptake | Histology, Immunohistochemistry |
| Zebrafish [15] | Oocyte heat stress protection | Lrp13 (VtgR) disruption | Impaired vitellogenin absorption; Ovarian degeneration at high temperatures | Morphological & histological analysis |
| Honey Bee (Apis mellifera) [2] | Division of labor & foraging specialization | RNAi (dsRNA injection) | Premature foraging onset; Nectar specialization; Reduced lifespan | Behavioral observation, Lifespan recording |
| Honey Bee (Apis mellifera) [5] | Swarming behavior | Gene expression analysis (qPCR) | Elevated Vg in nurse bees pre-swarming; Proposed role in colony reproduction | qRT-PCR on age-marked bees |
| Ant (Temnothorax longispinosus) [14] [16] | Behavioral task specialization | RNAi (fat body knockdown) | Reduced brood care; Increased nestmate care; Altered social cue responsiveness | Behavioral assays, Chemical cue tests |
| Melon Fly (Zeugodacus cucurbitae) [17] | Ovarian development | RNAi (dsRNA injection) | Significantly delayed ovarian development | Ovarian morphology & development staging |
| Kissing Bug (Rhodnius prolixus) [4] | Reproduction & Lifespan | RNAi (dsRNA injection) | Yolk-depleted eggs; Non-viable offspring; Increased lifespan in both sexes | Egg analysis, Survival assays |
Detailed Experimental Protocols for Key Findings
Protocol: RNAi-Mediated Vg Knockdown in Insects
The RNAi protocol has been successfully applied to study Vg function in honey bees, ants, and melon flies [2] [17] [14].
- dsRNA Preparation: Primers are designed with T7 promoter sequences from the target species' Vg cDNA sequence. The dsRNA is then synthesized in vitro [2].
- Delivery Method: Newly emerged adult insects are typically injected with a defined dose of dsRNA into the hemolymph or fat body. Controls receive injections of dsRNA for a non-target gene, such as green fluorescent protein (GFP) [2] [14].
- Validation of Knockdown: The efficiency of Vg knockdown is confirmed 5-7 days post-injection using:
- Phenotypic Observation: Treated individuals are monitored for behavioral changes (e.g., foraging onset, task specialization) or reproductive defects (e.g., oogenesis, egg viability) [2] [17].
Protocol: qRT-PCR Validation of Vg Knockdown
qRT-PCR is the standard method for confirming successful gene knockdown. A rigorous protocol is critical for reliable data [5] [18].
- RNA Extraction: Total RNA is isolated from the target tissue (typically abdomen or fat body) using commercial kits (e.g., Maxwell RSC SimplyRNA Tissue Kit). DNase treatment is included to remove genomic DNA contamination [5].
- cDNA Synthesis: Reverse transcription is performed on 1 µg of total RNA using a PrimeScript RT reagent kit [17].
- qPCR Amplification: Reactions are run in triplicate using a real-time PCR system (e.g., Bio-Rad CFX Connect). The reaction mix typically includes SYBR/FAM dye, primers for the target Vg gene, and primers for two validated reference genes [5].
- Data Analysis: The comparative ΔΔCt method is used to calculate relative gene expression. Data is normalized using the geometric mean of the two reference genes [5].
Essential Signaling Pathways Involving Vitellogenin
Vg functions within an intricate network of hormonal and signaling pathways, which explains its diverse physiological impacts.
- Reproductive Pathway: In most insects, Vg synthesis in the fat body is regulated by hormones. Juvenile hormone (JH) and ecdysone (20-hydroxyecdysone, 20E) act as key regulators, though their specific roles vary by species [17] [19]. In honey bees, Vg and JH exist in a unique double repressor network, where each suppresses the other, thereby influencing the transition from nursing to foraging behavior [2] [20].
- Insulin/IGF-1 Signaling (IIS) Pathway: This conserved pathway, which regulates aging, fertility, and metabolism, shows connections to Vg. In honey bee queens, high Vg expression correlates with low expression of insulin-like peptide (ILP) and its receptors in the head, suggesting an interaction that may contribute to their longevity [20].
- Antioxidant Pathway: Vg has demonstrated antioxidant capability. It can scavenge free radicals, which is hypothesized to reduce oxidative stress and thereby prolong lifespan in honey bee workers and queens [20].
The Scientist's Toolkit: Key Research Reagents & Materials
Table 2: Essential Reagents for Vg Knockdown and Validation Studies
| Reagent/Material | Function/Application | Example Use Case |
|---|---|---|
| dsRNA targeting Vg | Triggers sequence-specific mRNA degradation (RNAi) | Knockdown of Vg in honey bees, ants, and melon flies to study functional consequences [2] [17] [14]. |
| Vg Antibodies | Detect and quantify Vg protein levels via Western Blot or IHC | Confirmation of Vg protein knockdown and localization in tissues (e.g., mud crab oocytes) [15] [20]. |
| TRIzol Reagent | Monophasic solution for RNA isolation from cells/tissues | RNA extraction for downstream qRT-PCR analysis of Vg transcript levels [17]. |
| qRT-PCR Kits (One-Step or Two-Step) | Quantify gene expression levels from RNA samples | Validation of Vg knockdown efficiency and measurement of Vg expression under different conditions [17] [5]. |
| Validated Reference Genes | Stable internal controls for qRT-PCR normalization | Accurate normalization of Vg qPCR data; genes like β-actin, NDUFA8, H2A are used [5] [18]. |
| Hormones (JH, 20E) | Investigate hormonal regulation of Vg expression | Treatment studies to understand how hormones control Vg synthesis (e.g., in melon fly) [17]. |
The systematic comparison of Vg knockdown studies reveals a remarkable functional plasticity for this evolutionarily conserved protein. Beyond its canonical role in reproduction, Vg is a critical regulatory factor in thermal resilience, behavioral maturation, social organization, and lifespan. The consistency of phenotypic outcomes across distant taxa, when Vg is disrupted, underscores its fundamental importance. For researchers, this highlights the necessity of robust experimental design, including careful selection of reference genes for qRT-PCR and consideration of species-specific regulatory pathways, to accurately interpret the multifaceted roles of Vg in animal physiology and behavior.
Vitellogenin (Vg) is a glycolipoprotein that serves as a critical yolk precursor in oviparous animals. Beyond its fundamental role in reproduction, Vg has garnered significant research interest for its involvement in diverse physiological processes including aging, immune function, social behavior, and caste differentiation in social insects [21] [22] [23]. The establishment of robust, validated quantitative reverse transcription PCR (qRT-PCR) assays is paramount for accurate Vg gene expression quantification across these research contexts. This guide provides a comprehensive comparison of Vg study approaches and the essential validation requirements that ensure experimental reliability.
The context of use (COU) defines how a biomarker like Vg will be measured, the clinical or research purpose of these measurements, and how the results will be interpreted for decision-making [24]. Defining the COU is the critical first step that determines the appropriate level of validation rigor, adhering to the "fit-for-purpose" concept where validation stringency matches the intended application [24]. This framework guides researchers in selecting appropriate methodologies whether for basic research on insect physiology or developing clinical diagnostics.
Vg Gene Family Diversity and Functional Implications
Vitellogenin genes exhibit remarkable diversity across species, with multiple paralogs arising from gene duplication events. Phylogenetic analyses reveal that Vg and Vg-like genes cluster into distinct groups, with important functional implications. In the ant Temnothorax longispinosus, the studied Vg ortholog falls into a separate Vg-like A cluster rather than grouping with the intensively studied honey bee Vg [21]. This diversity necessitates species-specific assay validation and cautions against extrapolating functional annotations between distant taxa.
Table 1: Vitellogenin Gene Characteristics Across Species
| Species | Gene Name/Type | Key Functions | Expression Patterns |
|---|---|---|---|
| Honeybee (Apis mellifera) | Vitellogenin (Vg) | Royal jelly production, immunity, longevity, behavioral maturation [22] | Adult fat body, queen-specific brain glial cells [23] |
| Ant (Temnothorax longispinosus) | Vg-like A | Regulation of division of labor, social cue responsiveness [21] | Fat body, associated with brood care behavior [21] |
| Diamondback moth (Plutella xylostella) | VgR (Receptor) | Vg transport, yolk deposition, oocyte development [8] | Female-specific, predominantly in ovaries [8] |
The diagram below illustrates the core workflow for Vg gene expression studies and the key regulatory pathways involving vitellogenin:
Methodological Comparison for Vg Gene Manipulation
RNA Interference (RNAi) Approaches
RNAi has emerged as a powerful tool for probing Vg gene function in adult insects. Two primary delivery methods have been systematically compared for efficacy:
Table 2: RNAi Delivery Method Efficacy for Vg Knockdown
| Method | Efficiency | Persistence | Technical Complexity | Best Applications |
|---|---|---|---|---|
| Embryonic Microinjection (pre-blastoderm eggs) | 15% of adults showed mutant phenotype [22] | Detectable at emergence and persistent over 15 days [22] | High - requires precise embryonic manipulation | Studies where gene disruption in all developmental stages is acceptable |
| Intra-abdominal Injection (newly emerged adults) | 96% showed mutant phenotype [22] | RNA fragment present after 15 days; high molecular weight dsRNA persistent [22] | Moderate - adult injection simpler than embryonic work | Adult-specific gene function studies; when high penetrance is required |
The exceptional efficacy of intra-abdominal injection in honeybees may be attributed to the fat body's physiological role in uptaking macromolecules from hemolymph, analogous to the mammalian liver [22]. This method enables functional studies of Vg in adult bees without affecting developmental processes.
CRISPR/Cas9 Gene Editing
CRISPR/Cas9 technology provides a more permanent approach to Vg gene disruption. In the diamondback moth (Plutella xylostella), CRISPR-mediated knockout of the vitellogenin receptor (VgR) created homozygous mutants with a 5-bp nucleotide deletion, resulting in functional deficiencies [8]. The phenotypic consequences included:
- Shorter ovarioles in newly emerged females
- Smaller, whiter eggs with reduced hatching rates
- Decreased Vg protein expression in eggs despite unaffected Vg transcripts [8]
This approach demonstrates VgR's indispensable role in Vg transport and reproductive success, highlighting its potential as a genetic-based target for pest control strategies.
qRT-PCR Validation Framework for Vg Studies
Analytical Validation Parameters
qRT-PCR assays require rigorous validation to ensure data reliability. Key analytical performance characteristics must be established:
- Linearity and Range: Typically assessed using a 7-point 10-fold dilution series with R² ≥ 0.980 considered acceptable [25]
- Amplification Efficiency: Ideal range between 90-110% [18]
- Limit of Detection (LOD): The minimum detectable concentration (e.g., 0.003pg/reaction for Vero DNA assay) [26]
- Limit of Quantification (LOQ): The minimum quantifiable concentration (e.g., 0.03pg/reaction for Vero DNA assay) [26]
- Precision: Measured as relative standard deviation (RSD) across samples (e.g., 12.4-18.3%) [26]
Reference Gene Selection and Validation
Appropriate reference gene selection is critical for accurate Vg expression normalization. Studies must validate potential reference genes under specific experimental conditions, as stability can vary significantly:
Table 3: Reference Gene Stability Across Experimental Conditions
| Experimental Condition | Most Stable Reference Genes | Least Stable Reference Genes | Validation Method |
|---|---|---|---|
| Chemical exposure in Diaphanosoma celebensis | H2A, EF-1b, UBC, TBP, Act [18] | Atb, GAPDH [18] | GeNorm, NormFinder, BestKeeper, RefFinder |
| Different ages in Diaphanosoma celebensis | Different pattern from chemical exposure [18] | Significant variation with age [18] | GeNorm, NormFinder, BestKeeper, RefFinder |
| Various sweet potato tissues | IbACT, IbARF, IbCYC [27] | IbGAP, IbRPL, IbCOX [27] | RefFinder algorithm |
The striking difference in reference gene stability between chemical exposure and aging conditions in D. celebensis highlights the necessity of condition-specific validation rather than relying on conventional "housekeeping" genes without verification [18].
Experimental Protocols for Key Vg Studies
RNAi-Mediated Vg Knockdown in Social Insects
Protocol 1: Intra-abdominal dsRNA Injection for Adult Honeybees
- dsRNA Template Preparation: Design primers to amplify a 504 bp stretch of the Vg coding sequence [22]
- dsRNA Synthesis: Generate dsRNA using appropriate in vitro transcription systems
- Experimental Groups: Divide newly emerged bees into treatment (Vg-dsRNA) and control (unrelated dsRNA or buffer) groups
- Injection Procedure: Administer dsRNA solution intra-abdominally using microinjection apparatus
- Post-injection Incubation: Maintain injected bees in laboratory cages with appropriate feeding (e.g., 10% honey solution) [22]
- Sampling and Validation: Collect fat body tissue after 7 days for qRT-PCR analysis of Vg mRNA reduction
- Phenotypic Assessment: Monitor behavioral shifts, longevity, or immune function parameters based on research objectives
Validation Metrics: Successful knockdown should show >90% reduction in target mRNA, with minimal off-target effects confirmed through sequencing of observed RNA fragments [22].
qPCR Assay Validation for Regulatory Applications
Protocol 2: Validation for Residual DNA Detection in Biologics (Adaptable to Vg Studies)
- Target Selection: Identify unique, highly repetitive sequences specific to target species
- Primer/Probe Design: Design multiple amplicons (e.g., 99 bp and 154 bp fragments) to confirm sequence-independent detection [26]
- Linearity Assessment: Prepare 6-8 point 10-fold dilution series of standard DNA
- Precision Evaluation: Test inter- and intra-assay variability across multiple runs
- Specificity Testing: Verify absence of cross-reactivity with related species or common contaminants
- Robustness Determination: Assess performance under varying experimental conditions (e.g., different instruments, operators) [26]
This framework, developed for residual Vero DNA detection in rabies vaccines, provides a template for clinical-grade Vg assay validation [26].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagent Solutions for Vg Studies
| Reagent/Category | Specific Examples | Function/Application | Validation Requirements |
|---|---|---|---|
| RNAi Reagents | dsRNA targeting Vg coding sequence [22] | Gene knockdown studies; functional validation | Confirmation of mRNA reduction; phenotypic consistency |
| qPCR Master Mixes | Enzyme mixes, buffers, dNTPs, probes [26] | Target gene amplification and detection | Lot-to-lot consistency; minimal inhibition; stable fluorescence |
| Reference Genes | H2A, EF-1b, ACT, TBP [18] | Expression normalization | Stability validation under specific conditions [27] [18] |
| Nucleic Acid Standards | Vero genomic DNA standard [26] | Calibration curve generation | Certified concentration; sequence verification |
| cDNA Synthesis Kits | HiscriptTM Reverse Transcriptase [8] | RNA-to-cDNA conversion | Consistent efficiency; minimal degradation |
Establishing the appropriate context of use is foundational to Vg study design, determining whether research-use-only or clinically validated assays are required [24]. The methodological comparisons presented herein demonstrate that RNAi approaches offer high efficacy for adult functional studies, particularly in social insects where Vg influences complex behaviors [21] [22]. Meanwhile, CRISPR/Cas9 enables permanent genetic disruption for developmental and reproductive studies [8].
Robust qRT-PCR validation remains the cornerstone of reliable Vg quantification, requiring careful attention to reference gene stability [27] [18], amplification efficiency [25], and analytical specificity [26]. By aligning methodological rigor with research objectives through the fit-for-purpose framework [24], researchers can generate reproducible, biologically meaningful data that advances our understanding of vitellogenin's diverse roles across species and informs potential clinical applications.
Executing Vitellogenin Knockdown: RNAi Strategies and qRT-PCR Workflow
Designing Effective dsRNA for High-Penetrance Vg Silencing
Achieving high-penetrance silencing of genes such as vitellogenin (Vg) is a central goal in functional genetics and therapeutic development. The efficacy of RNA interference (RNAi) is profoundly influenced by the strategic design of the double-stranded RNA (dsRNA) trigger. A significant challenge in the broader context of qRT-PCR validation of vitellogenin knockdown research lies not just in inducing the silencing event, but in accurately measuring it. Research has demonstrated that inappropriate qRT-PCR primer design can lead to false-negative results and a significant underestimation of silencing efficiency, particularly for lowly expressed genes [28]. This guide objectively compares key dsRNA design and delivery parameters, providing supporting experimental data to help researchers navigate the path to robust and verifiable gene knockdown.
dsRNA Design Parameters: A Comparative Analysis
The structural and sequence-specific attributes of dsRNA are fundamental to its success. The table below compares critical design parameters based on empirical studies.
Table 1: Comparative Analysis of Key dsRNA Design Parameters
| Design Parameter | Approach 1 | Approach 2 | Experimental Support & Efficacy Data |
|---|---|---|---|
| Target Region Selection | Primers for qRT-PCR quantification bind within the dsRNA-targeted mRNA region. | Primers for qRT-PCR quantification bind outside the dsRNA-targeted mRNA region. | For low-expression genes, primers binding inside the region failed to show significant knockdown (false negative). Primers binding outside reliably detected significant knockdown [28]. |
| dsRNA Length | Short dsRNA (< 60 bp). | Long dsRNA (> 60 bp). | A size cut-off of ~60 bp is required for efficacy in insect models. Shorter dsRNAs (e.g., 21-bp siRNAs) showed no mortality, while longer dsRNAs resulted in high larval mortality [29]. |
| Targeting Strategy (Single vs. Multiple Genes) | dsRNA targeting a single essential gene. | dsRNA simultaneously targeting multiple genes (e.g., CYP3RNA targeting FgCYP51A, B, C). | Targeting multiple genes inhibited fungal growth more efficiently than targeting a single gene. Both strategies reduced infection, but single-gene knockdowns sometimes showed minimal phenotypic effect [30] [31]. |
dsRNA Delivery Methods: Efficacy and Workflows
The method of delivering dsRNA into the target organism is a critical determinant of silencing penetrance. The following table compares five common methods, with a subsequent diagram outlining a generalized experimental workflow.
Table 2: Comparison of dsRNA Delivery Methods for Silencing Efficacy
| Delivery Method | Description | Experimental Model | Key Efficacy Findings |
|---|---|---|---|
| Direct Soaking | Mites are soaked in a dsRNA solution. | Tetranychus urticae (spider mite) [32] | One of the most efficient methods, resulting in a clear dark-body phenotype and increased mortality/reduced fecundity. |
| Spray-Induced Gene Silencing (SIGS) | dsRNA is applied as a sprayable solution onto leaves. | Tetranychus urticae [32] & Fusarium graminearum [30] [31] | Highly efficient, mimicking a sprayable pesticide. Protects barley from fungal infection and induces strong phenotypes in mites. |
| Host-Induced Gene Silencing (HIGS) | dsRNA is constitutively expressed in transgenic host plants. | Arabidopsis, Barley [30] [31] & Tetranychus urticae [32] | Reduced fungal infection and mite fitness, though with variable efficiency. Offers continuous protection but involves transgenic plants. |
| Feeding on Coated Leaves | Leaves are coated with a layer of dsRNA. | Tetranychus urticae [32] | Highly efficient, comparable to direct soaking, and resulted in a dark-body phenotype. |
| Feeding on Artificial Diet | dsRNA is supplemented into an artificial liquid diet. | Bactericera cockerelli (potato psyllid) [33] | Ingestion of dsRNA targeting gut genes (e.g., AGLU1, AQP2) resulted in 20-60% gene knockdown and up to 40% mortality. |
The following diagram illustrates a generalized experimental workflow for designing, producing, and validating an effective dsRNA treatment, integrating the critical steps discussed in this guide.
Experimental Protocols for Key Methodologies
Protocol: dsRNA Delivery via Soaking and Spraying (SIGS)
This protocol is adapted from methods used to achieve high-efficacy silencing in spider mites and plants [32].
- dsRNA Synthesis: Synthesize dsRNA using a commercial in vitro transcription kit (e.g., MEGAscript RNAi Kit). The template should contain a T7 promoter sequence flanking the target gene fragment. Purify the resulting dsRNA.
- Delivery Setup:
- Soaking: Resuspend the target organism (e.g., mites, nymphs) in a solution containing 20% sucrose and dsRNA (e.g., 100-500 ng/μL). Allow an ingestion-access period (IAP) of 48 hours [33].
- Spraying (SIGS): Prepare an aqueous solution containing dsRNA. For potential field application, dsRNA can be complexed with nanocarriers like layered double hydroxide (LDH) clay nanosheets to enhance stability [30]. Apply the solution as a fine mist to fully cover the leaves of the host plant.
- Incubation and Monitoring: After delivery, transfer organisms to fresh, untreated host material. Monitor mortality, fecundity, and the emergence of specific phenotypic markers (e.g., dark-body phenotype in mites) over 3-9 days post-treatment [32] [33].
Protocol: qRT-PCR Validation with RNase If Treatment
Accurate quantification of intact mRNA post-RNAi requires distinguishing it from residual dsRNA and cleaved fragments. This modified qRT-PCR protocol is designed for this purpose [29].
- RNA Extraction: Extract total RNA from treated samples using a standard plant/fungi RNA purification kit. Include a DNase I treatment step to remove genomic DNA contamination.
- RNase If Treatment: Treat the extracted RNA with RNase If, a single-strand specific endonuclease. Under optimized high-salt conditions, RNase If preferentially degrades ssRNA (including mRNA and cleaved fragments) while leaving dsRNA intact.
- cDNA Synthesis & qPCR: Convert the RNase If-treated RNA into cDNA using a reverse transcription kit. Perform quantitative PCR using primer pairs designed to bind to the mRNA sequence extending beyond the region targeted by the dsRNA [28]. This ensures amplification only from intact, non-cleaved mRNA molecules, providing a true measure of knockdown efficiency.
The Scientist's Toolkit: Essential Reagents for dsRNA Research
Table 3: Key Research Reagent Solutions for dsRNA Experiments
| Research Reagent | Function / Application | Example Use Case |
|---|---|---|
| RNase If | An endonuclease that preferentially digests single-stranded RNA (ssRNA) over dsRNA. | Used in the RNase If-qPCR method to selectively remove ssRNA, allowing for precise quantification of dsRNA or intact mRNA in a sample [29]. |
| MEGAscript RNAi Kit | A commercial in vitro transcription kit for high-yield synthesis of dsRNA from a DNA template. | Used to produce large quantities of dsRNA for feeding, soaking, or spraying experiments [28]. |
| Norgen Plant/Fungi RNA Purification Kit | A commercial kit for the purification of high-quality total RNA from plant or fungal tissues. | Used to extract RNA from plant or insect samples post-dsRNA treatment for downstream qRT-PCR analysis [28]. |
| Ion-Pair Reverse Phase HPLC | A chromatographic method for the high-resolution analysis and purification of dsRNA. | Enables rapid purification of dsRNA from bacterial cell lysates and analysis of dsRNA integrity, separating it from contaminating DNA and ssRNA [34]. |
Designing dsRNA for high-penetrance vitellogenin silencing requires an integrated strategy that couples effective trigger design with a reliable validation methodology. The experimental data and comparisons presented herein demonstrate that employing long dsRNAs (>60 bp), considering multi-gene targeting strategies, and utilizing efficient delivery methods like soaking or SIGS, lay the foundation for potent knockdown. Crucially, this must be paired with a rigorous qRT-PCR protocol that uses primers positioned outside the dsRNA target region and, if necessary, an RNase If treatment step to avoid analytical pitfalls. By adopting this comprehensive approach, researchers can ensure that their measurements of Vg knockdown are both accurate and reflective of a true, high-penetrance silencing event.
In the field of molecular biology, particularly in research focused on gene function analysis such as vitellogenin knockdown studies, the selection of a delivery method for genetic materials is paramount. Two technically distinct approaches—intra-abdominal (intraperitoneal) injection and egg (in ovo or pronuclear) microinjection—offer researchers different pathways for introducing substances like dsRNA, CRISPR-Cas9 components, or therapeutic agents into their experimental models. Vitellogenin, a key yolk precursor protein critical for reproductive success in oviparous organisms, serves as a frequent target for gene knockdown experiments aimed at understanding reproductive biology, developmental processes, and pest control mechanisms. The validation of successful knockdown via quantitative real-time PCR (qRT-PCR) relies entirely on the efficiency and precision of the initial delivery method. This guide provides an objective comparison of these two techniques, framing their operational parameters, advantages, and limitations within the context of a vitellogenin knockdown research workflow, to assist researchers in selecting the most appropriate methodology for their specific experimental goals.
Intra-abdominal injection and egg microinjection represent fundamentally different approaches to substance delivery. The table below summarizes their core technical characteristics.
Table 1: Fundamental Characteristics of Intra-Abdominal Injection and Egg Microinjection
| Feature | Intra-Abdominal (Intraperitoneal) Injection | Egg (Microinjection) |
|---|---|---|
| Definition | Injection of a substance into the peritoneal (body) cavity [35]. | Direct injection of genetic material into a cell, typically a fertilized egg or embryo, using a fine glass needle [36] [37]. |
| Primary Applications | Commonly used for administering drugs, chemotherapy, and fluids in humans, and for delivering therapeutics in laboratory animals [35]. | Primarily used for creating transgenic animals, genome editing (e.g., CRISPR-Cas9), and intracytoplasmic sperm injection (ICSI) [36] [37]. |
| Standard Injection Volume | Variable, can accommodate larger fluid volumes [35]. | Very small, precise volumes (e.g., 0.5 mL into an avian egg [38]). |
| Technical Complexity | Relatively lower; a common laboratory procedure [35]. | High; requires specialized, expensive equipment and significant technical expertise [36] [37]. |
| Throughput | Suitable for administering treatments to multiple individual animals. | Low throughput; a laborious, single-cell-at-a-time process [36]. |
Figure 1: Conceptual Workflow of the Two Delivery Methods. Intra-abdominal injection targets the body cavity of developed organisms, while egg microinjection targets individual early embryonic cells.
Critical Experimental Performance Data
The choice between these methods is guided by hard data on their performance in experimental settings. The following tables consolidate key quantitative and qualitative outcomes from research contexts.
Table 2: Experimental Performance and Outcomes in Model Organisms
| Criterion | Intra-Abdominal (Intraperitoneal) Injection | Egg (Microinjection) |
|---|---|---|
| Efficiency in Gene Editing | Not typically used for this purpose. | High efficiency; one study reported 80-100% gene knockout in mammalian zygotes [36]. |
| Effect on Hatchability (Avian Eggs) | Not applicable. | Dose-dependent; 2.5% formula product concentration increased hatching rate, while 5% negatively impacted it [38]. |
| Impact on Intestinal Development | Not applicable. | Significant positive effect; a 2.5% formula product injection significantly increased villus height and crypt depth in chicks [38]. |
| Mortality & Toxicity | Potential for mis-injection and variability in effectiveness [35]. | Risk of cell damage and lysis; success highly dependent on operator skill [36] [37]. |
Table 3: Practical Research Considerations
| Criterion | Intra-Abdominal (Intraperitoneal) Injection | Egg (Microinjection) |
|---|---|---|
| Key Advantage | Can be used for large volumes and is suitable for systemic drug delivery [35]. | Precise control over the amount and location of delivered material; no carrier required [36] [37]. |
| Key Disadvantage | Variable absorption and potential for mis-injection, leading to inconsistent results [35]. | Low throughput, technically demanding, and can cause significant cell stress/damage [36] [37]. |
| Scalability | More easily scaled for studies requiring treatment of many post-natal animals. | Not scalable for large cell numbers; impractical for high-throughput applications [36] [37]. |
| Cost & Equipment | Lower cost; requires standard laboratory syringes and needles. | High cost; requires a micromanipulator, microinjector, micropipette puller, and a high-quality microscope [36] [37]. |
Application in Vitellogenin Knockdown: Experimental Protocols
The journey to successful qRT-PCR validation of vitellogenin (Vg) knockdown begins with the efficient delivery of silencing molecules, such as double-stranded RNA (dsRNA). The following protocols and data illustrate how the two methods are applied in a real research context targeting this key reproductive gene.
Intra-Abdominal Injection Protocol for Insect Vg Knockdown
While "intra-abdominal" is a broader term, in insects, injection into the body cavity (hemocoel) is a common procedure for delivering dsRNA. This protocol is adapted from studies involving the red palm weevil.
- Step 1: dsRNA Preparation: Synthesize and purify dsRNA targeting the Vg gene sequence. The dsRNA is resuspended in nuclease-free water [39].
- Step 2: Animal Preparation: Anaesthetize the insect (e.g., adult weevil) if necessary. The injection is typically performed on a cold surface to immobilize the subject.
- Step 3: Injection Process: Using a micro-syringe and a fine glass needle, the researcher punctures the intersegmental membrane of the cuticle, often in the abdomen, and delivers a defined volume (e.g., 10 μL) containing a specific dose (e.g., 4 μg) of Vg-dsRNA [39].
- Step 4: Post-Injection Care: The injected insect is transferred to optimal rearing conditions and monitored until sampling.
Egg Microinjection (In Ovo) Protocol for Avian Vg Studies
In ovo injection is a form of microinjection used in avian embryology to deliver substances directly into developing eggs.
- Step 1: Incubation and Candleing: Fertilized eggs are incubated until the desired developmental stage (e.g., day 18 of incubation for chicken eggs). Eggs are candled to identify viable embryos and mark the air sac [38] [40].
- Step 2: Preparation and Sterilization: The blunt end of the egg (over the air sac) is cleaned with 75% ethyl alcohol [38].
- Step 3: Piercing and Injection: A small hole is pierced in the shell. A sterile syringe with a 26G needle is used to inject the solution (e.g., 0.5 mL of a nutrient or dsRNA solution) into the air sac or amnion [38] [40]. The hole is then sealed with liquid paraffin or a glue.
- Step 4: Post-Injection Incubation: Injected eggs are returned to the incubator until hatching or the desired sampling stage.
Figure 2: Experimental Workflow for Vitellogenin Knockdown Studies. This diagram outlines the key steps from initial gene silencing delivery to final qRT-PCR validation, showing how phenotypic and molecular analyses converge.
Comparative Experimental Data in Vg Research
The effectiveness of these methods in Vg knockdown is demonstrated by concrete experimental results.
Table 4: Efficacy Data from Vitellogenin-Targeted Studies
| Delivery Method | Organism | Key Experimental Findings |
|---|---|---|
| dsRNA Injection (Body Cavity) | Red Palm Weevil (Rhynchophorus ferrugineus) | Injection of Vg-dsRNA successfully suppressed Vg gene function, leading to a significant decline in egg hatchability [39]. |
| Feeding (Oral dsRNA Delivery) | Red Palm Weevil (Rhynchophorus ferrugineus) | Delivery of Vg-dsRNA via drops (oral) also resulted in a significant decline in egg hatchability and Vg expression, though diet-incorporated dsRNA was less effective [39]. |
| RNAi (Feeding) | Nematode (Caenorhabditis elegans) | Knockdown of vitellogenin genes (e.g., vit-2, vit-5) via feeding with dsRNA-producing bacteria was successful, leading to measurable changes in lipid levels and lifespan [12]. |
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful execution of these delivery methods and subsequent qRT-PCR validation requires a suite of specialized reagents and tools. The following table details key solutions essential for this field of research.
Table 5: Essential Research Reagents and Materials for Delivery and Validation Experiments
| Item | Function/Application | Examples/Notes |
|---|---|---|
| dsRNA Synthesis Kit | To produce high-quality, template-specific double-stranded RNA for gene silencing experiments. | Critical for RNAi-mediated Vg knockdown. Requires a template from the target organism's Vg gene sequence [39]. |
| Micropipette Puller | To fabricate fine-tipped glass micropipettes for microinjection from glass capillaries. | Essential for microinjection; tip diameter is crucial for cell viability and injection efficiency [36] [37]. |
| Micromanipulator & Microinjector | To precisely position the micropipette and control the injection pressure/volume during microinjection. | A core component of the microinjection setup, allowing for sub-cellular precision [36] [37]. |
| Validated Reference Genes | Stable endogenous genes used for normalization in qRT-PCR to ensure accurate quantification of target gene (Vg) expression. | Selection is critical. Genes like H2A and pmp-3 have been validated as stable in some invertebrates under various conditions, unlike more variable genes like β-actin [18] [12]. |
| qRT-PCR Master Mix | A pre-mixed solution containing enzymes, dNTPs, and buffers optimized for sensitive and specific SYBR Green-based real-time PCR detection. | Typically includes reverse transcriptase for cDNA synthesis and a hot-start DNA polymerase for PCR amplification [18] [12]. |
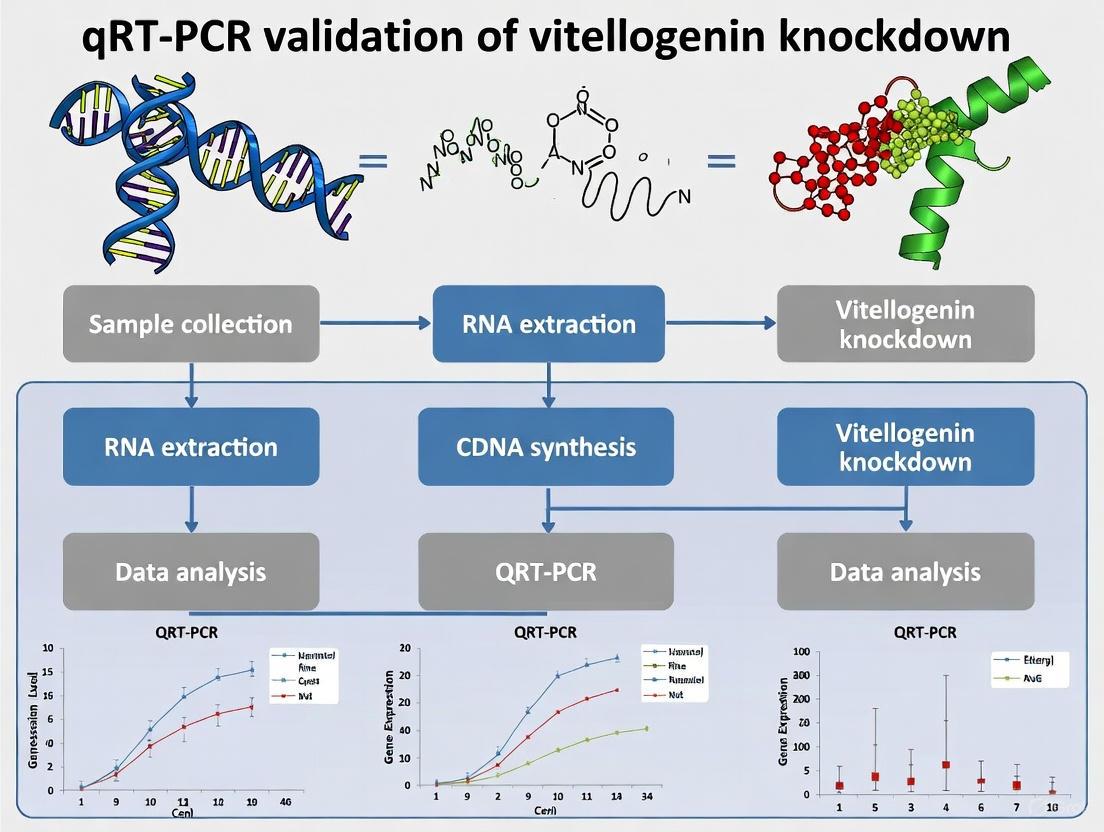
Quantitative reverse transcription polymerase chain reaction (qRT-PCR) serves as a fundamental technique in molecular biology for precisely measuring gene expression levels. This guide provides a comprehensive, step-by-step protocol for performing qRT-PCR, framed within the context of validating gene knockdown in research—specifically the knockdown of vitellogenin (vit-2) in C. elegans studies. We will objectively compare critical methodological choices and reagents based on experimental data, providing researchers with a reliable framework for generating publication-quality results.
Part 1: RNA Extraction and Quality Control
The foundation of any successful qRT-PCR experiment is high-quality, intact RNA.
Step 1: Sample Collection and Homogenization Rapidly collect tissue or cells of interest and immediately stabilize RNA using reagents like RNAlater or direct lysis in TRIzol. For C. elegans studies, synchronized animals are typically washed and collected in 1X PBS buffer before processing [12]. Flash-freeze samples in liquid nitrogen and store at -80°C if not processing immediately.
Step 2: RNA Isolation Use a commercially available column-based RNA extraction kit or a phenol-chloroform (e.g., TRIzol) method. For C. elegans, Tri-reagent has been effectively used according to the manufacturer's instructions [12]. Include a DNase I digestion step to remove contaminating genomic DNA.
Step 3: RNA Quantification and Quality Assessment Quantify RNA concentration using a spectrophotometer (NanoDrop) or fluorometer (Qubit). Assess RNA integrity via agarose gel electrophoresis (clear 18S and 28S ribosomal RNA bands) or using an instrument such as the Bioanalyzer. High-quality RNA should have an A260/A280 ratio between 1.8 and 2.1 and an RNA Integrity Number (RIN) greater than 8.0.
Part 2: cDNA Synthesis
This critical step reverse transcribes RNA into stable cDNA.
Step 4: Primer Selection Choose either oligo(dT) primers (for poly-A tailed mRNA), random hexamers (for broad coverage, including non-polyadenylated RNA), or gene-specific primers (for highest specificity but multiple reactions). For comprehensive expression profiling in C. elegans, oligo(dT) primers are commonly used, as in the SensiFAST cDNA synthesis kit [12].
Step 5: Reverse Transcription Reaction Set up the reaction on ice:
- Total RNA: 10 ng–1 µg (500 ng was used in vit-2 studies [12])
- Reverse Transcriptase: 1 µL (e.g., M-MLV, Superscript IV)
- Reaction Buffer: 1X concentration
- dNTPs: 0.5 mM each
- Primers: 2.5 µM oligo(dT) or 50 ng/µL random hexamers
- RNase Inhibitor: Optional, 10–20 Units
- Nuclease-free water: to 20 µL
Incubate as follows:
- 25°C for 10 min (primer annealing)
- 42–50°C for 30–60 min (elongation)
- 70°C for 15 min (enzyme inactivation)
Dilute the resulting cDNA 1:5 to 1:10 with nuclease-free water before use in qPCR.
Part 3: Quantitative PCR (qPCR) Setup and Amplification
Assay Design
Careful assay design is paramount for specificity and efficiency.
- Probe vs. SYBR Green: Probe-based qPCR (e.g., TaqMan) offers superior specificity and is recommended for regulatory studies due to reduced false-positive signals [41]. SYBR Green is more cost-effective but requires meticulous optimization and melt curve analysis to confirm specificity.
- Primer/Probe Design:
- Amplicon size: 80–150 bp
- Primer length: 18–22 bases
- Tm: 58–60°C (with <1°C difference between primers)
- Avoid runs of identical nucleotides and self-complementarity
- For probe design, ensure the Tm is 7–10°C higher than primers, avoid G at the 5' end, and place it close to the forward primer.
In vitellogenin knockdown research, specific primers and probes were designed for vit-2 and reference genes, with amplification efficiencies typically validated between 90% and 110% [12] [41].
Reaction Setup
A standard probe-based qPCR reaction mixture is detailed below.
Table 1: Probe-Based qPCR Reaction Setup
| Component | Final Concentration/Amount | Function |
|---|---|---|
| 2X TaqMan Master Mix | 1X | Provides DNA polymerase, dNTPs, buffer |
| Forward Primer | 300–900 nM [41] | Target-specific forward amplification |
| Reverse Primer | 300–900 nM [41] | Target-specific reverse amplification |
| TaqMan Probe | 50–300 nM [41] | Sequence-specific fluorescence detection |
| cDNA Template | 1–100 ng equivalent of input RNA | The target material for amplification |
| Nuclease-free Water | To final volume | Solvent |
Total Reaction Volume: 20–50 µL (a 50 µL volume was used in C. elegans vitellogenin studies [12])
Note: For SYBR Green-based reactions, replace the TaqMan Master Mix and probe with a SYBR Green Master Mix.
Plate and Instrument Selection
The choice of consumables can significantly impact data quality.
Table 2: qPCR Plate Selection Guide
| Plate Color | Best Use Case | Key Advantages | Limitations |
|---|---|---|---|
| White | qPCR/qRT-PCR | Maximizes fluorescence signal, reduces background noise, ideal for low-copy targets [42] [43] | More expensive than clear plates |
| Clear | Endpoint PCR, visualization | Low cost, easy sample visualization | Not ideal for fluorescence applications due to background interference [42] |
| Black | FRET assays, fluorescence microscopy | Reduces light reflection and cross-talk between wells [42] | Can absorb light, not optimal for standard qPCR |
For the most accurate and sensitive qRT-PCR results, white plates are strongly recommended as they reflect signal back to the detector, enhancing sensitivity and reducing well-to-well crosstalk [43].
Thermal Cycling
Run the plate on a real-time PCR instrument using a standard cycling protocol.
Table 3: Standard qPCR Thermal Cycling Conditions
| Stage | Temperature | Time | Cycles |
|---|---|---|---|
| Enzyme Activation | 95°C | 10 min | 1 |
| Denaturation | 95°C | 15 sec | 40 |
| Annealing/Extension | 60°C | 30–60 sec | 40 |
Data adapted from [41]. The annealing/extension time can be adjusted based on the polymerase used and amplicon length.
Part 4: Data Analysis
Step 6: Cycle Threshold (Ct) and Quantitation The Ct value is the cycle at which fluorescence crosses a threshold set in the exponential phase of amplification. For absolute quantitation, a standard curve with known copy numbers is required. The copy number in unknown samples is calculated using the formula [41]: $$ \text{DNA Quantity (copies)} = 10^{(\text{Ct value} - Y_{\text{inter}})/\text{slope}} $$
Step 7: Normalization to Reference Genes Normalize target gene data (e.g., vit-2) to stable reference genes to account for variations in input RNA and cDNA synthesis efficiency. Using at least two validated reference genes is critical [18]. In C. elegans lipid metabolism studies, pmp-3 has been used as a reference gene [12]. The most stable reference genes should be determined for your specific organism and experimental conditions [18].
Step 8: Calculation of Relative Expression The comparative ΔΔCt method is the most common way to calculate relative fold changes in gene expression between experimental and control groups.
Application in Vitellogenin Knockdown Research
The following diagram illustrates the experimental workflow and molecular pathway investigated in vitellogenin research, integrating the protocol steps within a biological context.
This experimental workflow was validated using the qRT-PCR protocol detailed in this guide. In the referenced study [12], knockdown of vit-1/2 via RNAi in adulthood resulted in a significant rescue of both lipid levels (almost 2-fold reduction in pry-1 mutants) and lifespan (102% increase in mean lifespan in pry-1 mutants), demonstrating the critical role of vit-2 downstream of pry-1.
Research Reagent Solutions
Table 4: Essential Reagents and Materials for qRT-PCR
| Item | Function | Example Products/Brands |
|---|---|---|
| RNA Extraction Kit | Isolate intact, pure total RNA | Tri-reagent (Sigma-Aldrich), QIAamp UCP Pathogen Mini Kit (Qiagen) [12] [44] |
| cDNA Synthesis Kit | Reverse transcribe RNA to cDNA | SensiFAST cDNA Synthesis Kit [12] |
| qPCR Master Mix | Provides enzymes and buffers for amplification | TaqPath ProAmp Master Mix (Thermo Fisher) [44], TaqMan Universal Master Mix II [41] |
| Sequence-Specific Primers/Probes | Target-specific amplification and detection | Custom designs from IDT [44] |
| Nuclease-Free Water | Solvent free of RNases and DNases | Various manufacturers |
| qPCR Plates | Hold reactions for thermal cycling and detection | Thermo Scientific white qPCR plates [43] |
| Optical Seals | Prevent evaporation and cross-contamination | Adhesive films or cap strips |
This step-by-step qRT-PCR protocol, from RNA extraction through data analysis, provides a robust framework for gene expression validation. When applied within the context of vitellogenin knockdown research, it effectively confirms the functional downstream role of vit-2 in lipid metabolism and lifespan regulation. By carefully selecting reagents, optimizing assay conditions, and employing rigorous normalization strategies, researchers can ensure the generation of precise, reproducible, and biologically meaningful data.
Selection and Validation of Stable Reference Genes for Normalization
Quantitative real-time polymerase chain reaction (qRT-PCR) represents the gold-standard technique for gene expression analysis due to its high sensitivity, specificity, and reproducibility [45]. However, its accuracy depends critically on stable reference genes for data normalization to account for technical variations introduced during RNA quality, cDNA synthesis efficiency, and PCR amplification [45] [46]. The selection of inappropriate reference genes can significantly distort gene expression profiles, leading to erroneous biological conclusions [46] [47]. This is particularly crucial in vitellogenin knockdown research, where accurately measuring subtle changes in gene expression is essential for validating knockdown efficiency and interpreting phenotypic outcomes.
Vitellogenin, a yolk protein precursor, plays critical roles in insect reproduction and embryo development [48]. Research across multiple insect species, including the red palm weevil (Rhynchophorus ferrugineus) and cotton boll weevil (Anthonomus grandis), has demonstrated that vitellogenin knockdown strongly affects egg viability and embryonic development [49] [48]. The normalization of vitellogenin expression data using properly validated reference genes is therefore fundamental to obtaining reliable results in reproductive biology studies.
Despite their importance, commonly used reference genes such as β-actin, GAPDH, and ribosomal proteins are not universally stable across different experimental conditions, tissues, or species [45] [18]. This article provides a comprehensive comparison guide for selecting and validating stable reference genes, with specific application to vitellogenin knockdown research in various biological models.
The Critical Role of Reference Genes in Gene Expression Studies
Why Reference Gene Validation Matters
Reference genes, often called "housekeeping genes," are presumed to maintain consistent expression across various experimental conditions. However, numerous studies have demonstrated that this presumption is often false, as the expression stability of these genes varies significantly depending on experimental treatments, tissue types, developmental stages, and species [18] [47]. The use of unvalidated reference genes can lead to substantial errors in gene expression quantification—in some cases producing completely opposite biological interpretations [46].
In vitellogenin research, proper normalization is particularly important because:
- Vitellogenin expression levels can vary dramatically during development and between tissues
- Knockdown experiments often produce partial rather than complete silencing
- Small changes in expression can have significant biological consequences for reproduction
- Different delivery methods for dsRNA (injection vs. feeding) may require different normalization strategies [49]
Consequences of Improper Normalization
The impact of inappropriate reference gene selection is well-documented across multiple studies. In cancer research, the expression of commonly used reference genes like ACTB, RPS23, RPS18, and RPL13A undergoes dramatic changes in dormant cancer cells treated with mTOR inhibitors, potentially distorting gene expression profiles if used for normalization [46]. Similarly, in the brackish water flea (Diaphanosoma celebensis), reference gene stability varies significantly between chemical exposures and different developmental ages, affecting the expression patterns of development and detoxification-related genes [18].
In functional validation experiments using the odorant receptor gene StriOR20 in Scotogramma trifolii, significant discrepancies in relative expression levels occurred when normalization was performed with unstable versus stable reference genes, emphasizing the necessity of rigorous reference gene selection [45].
Methodologies for Reference Gene Validation
Experimental Design and Sample Collection
Proper experimental design is fundamental for reference gene validation. Samples should represent the entire range of experimental conditions expected in subsequent studies. For vitellogenin knockdown research, this typically includes:
- Multiple developmental stages: Eggs, larvae, pupae, and adults, as vitellogenin expression is often developmentally regulated [45] [48]
- Various tissues: Fat body, ovary, and other reproductive tissues where vitellogenin is synthesized or accumulated [45]
- Treatment conditions: Both control and experimental groups, including dsRNA-treated and untreated individuals [49] [48]
Each sample category should include adequate biological replicates (typically 3-5) to account for natural variation [45]. For example, in Scotogramma trifolii research, five biological replicates were established for each sample category across four developmental stages and six adult tissues [45].
RNA Extraction and cDNA Synthesis
High-quality RNA is essential for reliable qRT-PCR results. Standard protocols include:
- RNA extraction: Using commercial kits (e.g., TransZol Up Plus RNA Kit, TRIzol reagent) following manufacturer protocols [45] [48]
- Quality assessment: Measuring RNA concentration and purity using spectrophotometry (A260/280 ratios of 1.8-2.2 indicate pure RNA) [45] [47]
- DNA removal: Treating with DNase I to eliminate genomic DNA contamination [48]
- cDNA synthesis: Using 0.5-1 μg of total RNA with reverse transcriptase and oligo(dT) or random primers [45] [12]
Candidate Reference Gene Selection
Candidate reference genes are typically selected from two main categories:
- Traditional housekeeping genes: Involved in basic cellular maintenance (e.g., GAPDH, ACTB, TUB)
- Less conventional genes: Identified from transcriptomic datasets as having stable expression [50]
The number of candidate genes varies across studies, but typically ranges from 6 to 14 genes [45] [50] [47]. For example, in nasturtium (Tropaeolum majus), 14 candidate genes were evaluated from transcriptome data [50].
Primer Design and Validation
Proper primer design is crucial for specific and efficient amplification:
- Design parameters: Primer length of 18-25 bp, amplicon size of 80-200 bp, GC content of 40-60%, and annealing temperature of 58-62°C [47]
- Specificity verification: Using conventional PCR and agarose gel electrophoresis to confirm single bands of expected size [51]
- Efficiency testing: Creating standard curves with serial cDNA dilutions; amplification efficiency of 90-110% is generally acceptable [18] [51]
- Melting curve analysis: Conducting post-amplification melting curves to confirm single products [51]
Stability Analysis Algorithms
Four algorithmic tools are commonly used to evaluate reference gene stability:
- geNorm: Determines the most stable genes by calculating the average pairwise variation between genes [45]
- NormFinder: Uses an ANOVA-based model to estimate intra- and inter-group variations [45]
- BestKeeper: Relies on raw Ct values to calculate standard deviations and correlations [45]
- RefFinder: Integrates results from geNorm, NormFinder, BestKeeper, and the comparative ΔCt method to provide a comprehensive ranking [45] [18]
Figure 1: Workflow for reference gene selection and validation. The process begins with RNA extraction and proceeds through multiple analytical steps to identify the most stable reference genes for specific experimental conditions.
Comparative Analysis of Reference Genes Across Species and Conditions
Reference Gene Performance in Insect Models
Insect vitellogenin research has benefited from systematic reference gene validation across multiple species:
Table 1: Stable Reference Genes in Insect Species
| Species | Experimental Conditions | Most Stable Reference Genes | Least Stable Reference Genes | Functional Validation |
|---|---|---|---|---|
| Scotogramma trifolii (Clover cutworm) | Developmental stages | β-actin, RPL9, GAPDH | TUB, RPL9 (in some tissues) | Odorant receptor gene StriOR20 [45] |
| Scotogramma trifolii (Clover cutworm) | Adult tissues | RPL10, GAPDH, TUB | β-actin, EF1-α | Odorant receptor gene StriOR20 [45] |
| Diaphanosoma celebensis (Brackish water flea) | Chemical exposure (B[a]P, BPA, Hg) | H2A, EF-1b, UBC, TBP | Atb, GAPDH | EcRA and GST genes [18] |
| Diaphanosoma celebensis (Brackish water flea) | Different ages (24h-10 days) | Different pattern from chemical exposure | Significant variation with age | EcRA and GST genes [18] |
| Rhynchophorus ferrugineus (Red palm weevil) | Vg dsRNA feeding | Not specified in study | Not specified in study | Vitellogenin expression [49] |
The table demonstrates that optimal reference genes are highly condition-specific, underscoring the necessity for validation in each experimental system.
Reference Gene Performance in Non-Insect Models
Reference gene stability has been extensively studied in diverse biological systems:
Table 2: Stable Reference Genes in Non-Insect Systems
| Species/System | Experimental Conditions | Most Stable Reference Genes | Least Stable Reference Genes | Validation Approach |
|---|---|---|---|---|
| Tropaeolum majus (Nasturtium) | Different organs | EXP1, EXP2, TUB6 | Variable across conditions | KCS11 (fatty acid elongase) [50] |
| Tropaeolum majus (Nasturtium) | Seeds at different development stages | EXP1, CYP2 | Variable across conditions | KCS11 (fatty acid elongase) [50] |
| Inonotus obliquus (Fungus) | Different carbon sources | VPS | Variable across conditions | Not specified [51] |
| Inonotus obliquus (Fungus) | Different nitrogen sources | RPB2 | Variable across conditions | Not specified [51] |
| Inonotus obliquus (Fungus) | Different growth stages | VAS | Variable across conditions | Not specified [51] |
| Cancer cell lines (A549, T98G, PA-1) | mTOR inhibition (dormant cells) | B2M, YWHAZ (A549); TUBA1A, GAPDH (T98G) | ACTB, RPS23, RPS18, RPL13A | Not specified [46] |
| Kengyilia melanthera (Plant) | Abiotic stresses (drought, heat, cold, salt, ABA) | CACS, PPP2R1B (overall); TCTP, TIPRL (ABA) | Variable across stresses | Catalase-1 (CAT1) gene [47] |
Impact of Experimental Conditions on Reference Gene Stability
The stability of reference genes is significantly influenced by experimental conditions:
- Chemical exposures: In Diaphanosoma celebensis, optimal reference genes differed when exposed to B[a]P (Act and GAPDH) versus mercury (UBC and TBP) [18]
- Developmental stages: In Scotogramma trifolii, different reference genes were optimal for developmental stages (β-actin, RPL9, GAPDH) versus adult tissues (RPL10, GAPDH, TUB) [45]
- Environmental stresses: In Kengyilia melanthera, the most stable reference genes varied across drought (CACS, FBXO6L), heat (CACS, FBXO6L), cold (CACS, TCTP), salt (TIPRL, CYPA3), and ABA (TCTP, TIPRL) treatments [47]
- Cellular states: In dormant cancer cells, commonly used reference genes like ACTB and ribosomal proteins were categorically inappropriate after mTOR inhibition [46]
Figure 2: Impact of experimental conditions on reference gene stability. Different experimental conditions significantly affect which reference genes are most stable, necessitating condition-specific validation.
Application to Vitellogenin Knockdown Research
Vitellogenin Function and Research Significance
Vitellogenin is a phospholipoglycoprotein that serves as the primary yolk protein precursor in oviparous animals, including insects [48]. It is synthesized in the fat body, secreted into hemolymph, and transported to the ovary, where it is internalized into oocytes and processed into vitellin, the major nutrient source for embryo development [48]. Beyond its role in reproduction, vitellogenin has been implicated in foraging behavior, hormonal dynamics, immune response, and oxidative stress resistance in various insects [48].
Vitellogenin knockdown research has significant implications for:
- Basic science: Understanding reproductive biology and embryo development
- Pest management: Developing targeted control strategies for agricultural pests [49] [48]
- Biotechnology: Engineering disease resistance in beneficial insects
Reference Gene Selection for Vitellogenin Studies
Based on comparative analysis across multiple studies, the following guidelines emerge for vitellogenin knockdown research:
Multi-tissue studies: When analyzing vitellogenin expression across multiple tissues, validate reference genes specifically for each tissue type. In Scotogramma trifolii, different reference genes were optimal for different adult tissues [45]
Developmental studies: When examining vitellogenin expression across developmental stages, select reference genes validated for developmental stability. Vitellogenin expression typically varies dramatically during development [48]
dsRNA treatment studies: When using dsRNA delivery methods (injection or feeding), validate reference genes under the specific treatment conditions, as cellular stress responses might affect reference gene stability
Species-specific validation: Always perform initial validation for your specific species, as reference gene stability shows species-specific patterns [45] [18]
Experimental Design for Vitellogenin Knockdown Studies
Proper experimental design for vitellogenin knockdown research should include:
- Appropriate controls: Untreated and empty-vector/dsRNA controls
- Multiple time points: To capture dynamics of knockdown efficiency
- Tissue-specific analysis: Fat body, ovary, and hemolymph for comprehensive understanding
- Biological replicates: Minimum of 3-5 replicates to account for individual variation
- Functional validation: Include phenotypic measurements (egg production, viability) alongside molecular analyses [49] [48]
Essential Research Reagents and Tools
Table 3: Research Reagent Solutions for Reference Gene Validation
| Reagent/Tool Category | Specific Examples | Function/Application | Considerations |
|---|---|---|---|
| RNA Extraction Kits | TransZol Up Plus RNA Kit, TRIzol reagent, RNeasy Micro Kit, Monad RNA extraction kit | High-quality RNA isolation from diverse sample types | Assess yield and purity; check for genomic DNA contamination [45] [47] [48] |
| cDNA Synthesis Kits | EasyScript One-Step gDNA Removal and cDNA Synthesis SuperMix, SensiFAST cDNA Kit, Evo M-MLV RT Mix Kit, Hifair III 1st Strand cDNA Synthesis Kit | Efficient reverse transcription with genomic DNA removal | Use consistent RNA input amounts; include no-RT controls [45] [12] [47] |
| qPCR Master Mixes | SensiFAST SYBR Green Kit, Hieff qPCR SYBR Green Master Mix, SYBR Green ROX Plus PCR Mix | Sensitive detection with minimal background | Verify compatibility with detection system; optimize concentrations [12] [51] [48] |
| Primer Design Tools | Primer Premier, Beacon Designer, Primer-BLAST | Specific primer design with appropriate parameters | Check specificity; verify amplification efficiency [45] [50] [47] |
| Stability Analysis Software | geNorm, NormFinder, BestKeeper, RefFinder | Comprehensive evaluation of reference gene stability | Use multiple algorithms for robust conclusions [45] [18] [47] |
| qPCR Instruments | NanoDrop spectrophotometer, QuantStudio systems, BioRad CFX systems, ViiA7 systems | Accurate quantification and detection | Regular calibration; maintain consistent thermal cycling conditions [45] [50] [51] |
The selection and validation of stable reference genes is a critical prerequisite for reliable vitellogenin knockdown research and gene expression studies in general. As demonstrated by comparative analyses across multiple species and experimental conditions, no universal reference genes exist that perform optimally in all contexts. The most stable reference genes vary significantly across species, tissues, developmental stages, and experimental treatments.
Researchers investigating vitellogenin function should prioritize the validation of reference genes specifically tailored to their experimental systems, using multiple algorithms for stability analysis and including functional validation with target genes. By adopting these rigorous approaches, the scientific community can ensure the accuracy and reproducibility of vitellogenin expression data, advancing our understanding of reproductive biology and facilitating the development of novel pest management strategies.
The experimental protocols and comparative data presented in this guide provide a foundation for designing robust reference gene validation strategies in vitellogenin research and related fields. As qRT-PCR technologies continue to evolve, the principles of proper normalization remain fundamental to generating meaningful biological insights.
Vitellogenin (Vg) is a highly conserved yolk precursor protein essential for reproduction in most oviparous species. It is synthesized in extra-ovarian tissues (such as the fat body in insects or hepatopancreas in crustaceans), transported through the hemolymph or blood, and absorbed by developing oocytes via receptor-mediated endocytosis to provide nutritional support for embryonic development [15] [52] [7]. The critical role of Vg in reproduction makes it a promising target for genetic control strategies in pest species and a valuable model for studying gene function in non-model organisms. Assessing the efficacy of Vg knockdown requires a multifaceted approach that quantifies reduction at both transcriptional (mRNA) and translational (protein) levels, and crucially, connects these molecular changes to phenotypic outcomes in reproduction. This guide synthesizes experimental data and methodologies from recent studies to objectively compare measurement approaches and their applications in validation workflows, with particular focus on qRT-PCR as the cornerstone technique for mRNA quantification.
Methodological Approaches for Knockdown Validation
qRT-PCR: The Gold Standard for mRNA Quantification
Quantitative real-time polymerase chain reaction (qRT-PCR) serves as the primary method for directly quantifying changes in Vg mRNA expression following knockdown treatments. This technique provides high sensitivity, specificity, and quantitative accuracy for measuring transcript abundance.
- Experimental Workflow: Total RNA is extracted from target tissues (typically fat body or hepatopancreas), reverse-transcribed into cDNA, and amplified using gene-specific primers with fluorescent detection. The 2^(-ΔΔCt) method is commonly employed to calculate fold-changes in gene expression relative to control groups and housekeeping genes [53] [7].
- Key Considerations: RNA quality and integrity are critical factors. Studies specify using samples with RNA Integrity Number (RIN) >7 and DV200 values >15% to ensure reliable results [54] [55]. Proper normalization requires validation of stable reference genes under experimental conditions.
Complementary Techniques for Protein and Phenotypic Assessment
While qRT-PCR measures transcriptional effects, comprehensive validation requires complementary techniques to confirm functional protein reduction and physiological consequences.
- Western Blotting and SDS-PAGE: These techniques detect changes in Vg protein levels following knockdown. For example, research on Rhynchophorus ferrugineus demonstrated "dramatic failure of Vg protein expression" post-knockdown using SDS-PAGE analysis [7].
- Immunohistochemical Staining: Provides spatial localization of Vg protein within tissues. A study in mud crabs used Vg antibody staining to show absent signals in oocytes of abnormal crabs with impaired Vg uptake [15].
- Phenotypic Validation: Ultimate confirmation of functional knockdown comes from documenting defective oogenesis, reduced fecundity, and abnormal ovarian development through histological examination and reproductive assays [56] [7].
Table 1: Comparison of Vitellogenin Knockdown Efficacy Across Species
| Organism | Knockdown Method | mRNA Reduction | Protein/Phenotypic Effects | Experimental Duration |
|---|---|---|---|---|
| Red Palm Weevil (Rhynchophorus ferrugineus) [7] | dsRNA injection | 95-99% reduction (15-25 days) | Dramatic Vg protein failure; impaired oogenesis | 15-25 days |
| Red Imported Fire Ant (Solenopsis invicta) [56] | dsRNA injection | Significant downregulation (qRT-PCR) | Smaller ovaries; reduced egg production | 2-3 days |
| Lady Beetle (Coccinella septempunctata) [55] | dsRNA injection | Significant downregulation (RNA-seq & qRT-PCR) | Blocked vitellogenesis; repressed ovarian growth | 2 days |
| Mud Crab (Scylla paramamosain) [15] | Natural mutation (VgR enhancer) | Low VgR expression | Impaired Vg absorption; ovarian degeneration | Natural model |
Advanced Quantification Approaches
More sophisticated approaches have been developed for challenging targets:
- Multiplex qPCR Arrays: Enable simultaneous quantification of multiple gene targets. A 10-gene multiplex assay for bladder cancer research demonstrated robustness across RNA input levels (5-100 ng), different tissue types (FFPE and fresh-frozen), and minimal interference from necrosis [54].
- CircRNA Validation: For circular RNA targets, adding reverse primers to reverse transcription reactions improves reproducibility and accuracy of qRT-PCR despite low expression levels (1/10 to 1/1000 of their linear counterparts) [57].
Quantitative Comparison of Knockdown Efficacy Across Models
The efficacy of Vg knockdown varies substantially depending on the delivery method, organism, and timepoint of assessment. The compiled data from recent studies enables evidence-based comparison of expected outcomes.
Table 2: Temporal Progression of Knockdown Effects on Vg Expression
| Time Post-Treatment | Molecular-Level Effects | Cellular/Tissue Effects | Organismal Phenotypes |
|---|---|---|---|
| 2-3 days | Significant Vg mRNA downregulation [55] | Initial suppression of vitellogenesis [55] | Not yet evident |
| 15-20 days | 95-96.6% mRNA reduction [7] | Dramatic Vg protein failure [7] | Impaired oogenesis [7] |
| 25+ days | Up to 99% mRNA reduction [7] | Atrophied ovaries [7] | Complete cessation of egg hatchability [7] |
Experimental Protocols for Knockdown Validation
RNA Interference and qRT-PCR Validation Protocol
The following methodology is compiled from multiple studies demonstrating successful Vg knockdown validation [7] [55]:
- dsRNA Preparation: Design dsRNA targeting a unique region of the Vg transcript (300-500 bp). Use specific primers with appended T7 promoter sequences for in vitro transcription. Purify dsRNA using standard molecular biology techniques.
- Delivery: For insects, inject 1μL of dsRNA solution (2μg/μL) into the hemolymph through internode membranes between abdominal segments. Include control groups receiving dsRNA targeting irrelevant genes (e.g., GFP).
- Sample Collection: At appropriate timepoints post-injection (e.g., 2, 15, 25 days), dissect target tissues (fat body, hepatopancreas, or ovaries). Divide samples for RNA and protein extraction.
- RNA Extraction and qRT-PCR: Extract total RNA using TRIzol reagent. Assess RNA quality (RIN >7, DV200 >15%). Synthesize cDNA and perform qRT-PCR with validated reference genes. Calculate fold-change using the 2^(-ΔΔCt) method.
- Protein Analysis: Confirm reduced Vg translation via Western blot or SDS-PAGE of hemolymph or ovarian extracts.
- Phenotypic Assessment: Document ovarian development defects through histological staining and measure fecundity (egg production) and fertility (egg hatchability).
Vg Signaling and Knockdown Impact Pathway
The diagram below illustrates the vitellogenin synthesis, transport, and uptake pathway, with key knockdown targets highlighted.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Vg Knockdown Studies
| Reagent/Material | Function/Application | Example Specifications |
|---|---|---|
| dsRNA Synthesis Kit | Production of dsRNA for RNAi | MEGAscript T7 High Yield Transcription Kit [55] |
| RNA Extraction Reagent | Isolation of high-quality total RNA | TRIzol Reagent [55] |
| qRT-PCR Master Mix | Quantitative mRNA detection | SYBR Green or TaqMan chemistries [53] [54] |
| Vg-Specific Antibodies | Protein-level validation via Western blot/IHC | Species-specific Vg antibodies [15] [7] |
| Reference Genes | qRT-PCR normalization | Validated housekeeping genes (e.g., Tubulin) [7] |
| RNA Quality Assessment | Ensure RNA integrity | RIN >7, DV200 >15% [54] [55] |
Robust assessment of Vg knockdown efficacy requires an integrated approach combining qRT-PCR for precise mRNA quantification with protein-level analysis and phenotypic validation. The experimental data compiled in this guide demonstrates that effective Vg knockdown typically achieves >90% reduction at the mRNA level, leading to profound defects in oogenesis and reproductive failure. The methodologies outlined provide researchers with a standardized framework for comparing knockdown efficacy across different experimental systems, with qRT-PCR serving as an essential, but not standalone, component of a comprehensive validation strategy. As research in this field advances, the development of more sensitive multiplex assays and standardized protocols will further enhance our ability to precisely quantify gene knockdown and its functional consequences.
Troubleshooting qRT-PCR Assays: Overcoming Pitfalls for Robust Vg Quantification
In quantitative real-time PCR (qRT-PCR) research, particularly in sensitive applications like vitellogenin knockdown validation, optimal primer and probe design forms the foundational step that determines experimental success. These oligonucleotides must achieve perfect synergy: primers must efficiently amplify the specific target sequence, while probes must accurately report its presence, all while avoiding structural pitfalls that compromise data integrity. The challenge intensifies when working with genes like vitellogenin, which often have family members with high sequence similarity, increasing the risk of non-specific amplification. Secondary structures such as hairpins and primer-dimers represent the most common design failures, leading to inefficient reactions, false positives, and inaccurate quantification [58] [59]. This guide objectively compares design strategies and tools, providing researchers with methodologies to systematically avoid these pitfalls, ensuring that results in vitellogenin expression studies truly reflect biological reality rather than design artifacts.
Fundamental Principles of Primer and Probe Design
Core Design Parameters for Optimal Performance
Successful qRT-PCR assays rely on oligonucleotides that meet specific thermodynamic and compositional criteria. These parameters ensure efficient, specific amplification and detection while minimizing non-specific interactions.
Length and Melting Temperature (Tm): PCR primers should be 18-30 nucleotides long, with an optimal Tm of 60-64°C. The Tm difference between forward and reverse primers should not exceed 2°C. For TaqMan probes, the Tm should be 5-10°C higher than the primers, typically achieved with lengths of 20-30 nucleotides [60]. This Tm differential ensures the probe hybridizes before the primers, enabling efficient 5' nuclease cleavage [60].
GC Content and Sequence Composition: The GC content for both primers and probes should ideally be between 40-60% [59] [60]. This provides sufficient sequence complexity without promoting overly stable bonding. A "GC clamp"—the presence of one or two G or C bases at the 3' end of primers—can enhance specific binding, but more than three can cause non-specific annealing [59]. Avoid runs of four or more identical nucleotides, especially G residues [60].
Specificity and Genomic DNA Exclusion: To prevent amplification of genomic DNA, design primers to span an exon-exon junction [58]. This ensures amplification only from spliced mRNA. Furthermore, always run a minus-reverse transcriptase control ("No Amplification Control" or NAC) to detect genomic DNA contamination [58].
The Critical Threat: Secondary Structures and Primer-Dimers
Secondary structures refer to undesirable intramolecular or intermolecular interactions that outcompete target binding, severely reducing amplification efficiency and quantitative accuracy.
Hairpins (Self-Complementarity): These form when a primer or probe folds onto itself, creating a stable internal loop. This prevents the oligonucleotide from hybridizing to its target. Hairpins with low ΔG values (more negative than -9 kcal/mol) are particularly problematic [60].
Self-Dimers and Cross-Dimers: Self-dimers occur when two identical primers hybridize to each other. Cross-dimers form between forward and reverse primers. Like hairpins, these interactions sequester primers from the reaction. Primer-dimer is a common cause of false-positive amplification in no-template controls and can become the dominant amplification product, consuming reaction components [59].
The following workflow illustrates a systematic approach to designing and validating primers and probes that avoid these structures.
Comparative Analysis of Primer and Probe Design Tools
Various software tools are available to assist researchers in implementing these design principles. The table below provides an objective comparison of leading platforms.
Table 1: Comparison of qPCR Primer and Probe Design Tools
| Tool Name | Provider | Key Features | Strengths | Considerations |
|---|---|---|---|---|
| PrimerQuest | IDT | Customizes ~45 parameters; algorithm reduces primer-dimer formation; integrated BLAST analysis [61] [60]. | Highly customizable; user-friendly interface; direct ordering integration. | Vendor-associated; advanced features may have a learning curve. |
| Primer-BLAST | NCBI | Combines Primer3 design with BLAST specificity validation; options to span exon junctions [62]. | Unparalleled specificity checking; freely accessible; high confidence in unique targets. | Can be slower due to comprehensive BLAST search. |
| qPCR Assay Design Tool | Eurofins Genomics | Based on Prime+ (GCG Wisconsin Package); considers probe Tm 8-10°C higher than primer Ta [63]. | Robust parameter set; automatically avoids 5' G in probes. | Less publicly documented algorithm compared to open-source tools. |
| Real-time PCR Design Tool | GenScript | Designs TaqMan primers and probes; allows manual specification of exon junctions with ':' in sequence [64]. | Simple interface for standard assays; quick results. | Less customizable than other tools; requires login. |
Experimental Protocols for Validation
In Silico Analysis and Specificity Verification
Before synthesizing oligonucleotides, comprehensive computational checks are essential.
- Sequence Input: Input the exact target mRNA sequence (e.g., vitellogenin transcript from a model organism) into your chosen design tool. For C. elegans vitellogenin genes (vit-1 to vit-6), use RefSeq accessions for accuracy [12] [62].
- Parameter Setting: Apply the standard parameters from Table 1. For genes with high homology (like vit genes), increase stringency for 3'-end complementarity and overall mismatch tolerance.
- Secondary Structure Analysis: Use tools like IDT's OligoAnalyzer to input candidate sequences. Examine the results for:
- Specificity Check (BLAST): Run a NCBI BLAST alignment for each primer and the predicted amplicon against the appropriate genome (e.g., C. elegans) to ensure they are unique to the intended vitellogenin target and do not amplify other vit family members or non-target genes [60].
Wet-Lab Validation of Designed Assays
Even the best in-silico design requires empirical validation. The following protocol, adapted from vitellogenin knockdown studies, ensures the assay is efficient and specific [12].
Efficiency Curve:
- Prepare a serial dilution (e.g., 1:10, 1:100, 1:1000) of cDNA synthesized from a sample expressing the target vitellogenin gene.
- Run the qPCR assay with these dilutions.
- Plot the Log10 of the dilution factor against the Ct value. The slope of the line is used to calculate efficiency: Eff = 10(–1/slope) – 1. An ideal reaction efficiency is 90-110% (slope between -3.6 and -3.1) [58].
Analysis of Amplification and Melting Curves:
- Amplicon Specificity: For SYBR Green assays, run a dissociation (melting) curve at the end of the PCR. A single sharp peak indicates specific amplification of a single product. Multiple peaks suggest non-specific products or primer-dimer [58].
- Baseline and Threshold Setting: Set the baseline two cycles earlier than the Ct value for the most abundant sample. The threshold should be set in the exponential phase of amplification, typically at least 10 standard deviations above the baseline [58].
Critical Controls:
- No Template Control (NTC): Contains all reagents except RNA/cDNA. Used to rule out contamination of reagents with amplicon or primer-dimer formation [58].
- No Amplification Control (NAC): Contains all reagents except reverse transcriptase. Used to detect amplification from contaminating genomic DNA [58].
The relationship between optimal design, experimental validation, and reliable gene expression data is summarized in the following pathway.
Application in Vitellogenin Knockdown Research
In a study investigating the role of vit-2 in C. elegans lipid regulation downstream of PRY-1/Axin, researchers successfully applied these rigorous design principles [12]. The primers for vit-2 (FP: GACACCGAGCTCATCCGCCCA, RP: TTCCTTCTCTCCATTGACCT) and other vit genes were designed to be specific despite high sequence similarity among family members. The use of a reference gene, pmp-3, which was validated for stable expression, allowed for accurate normalization of qRT-PCR data during RNAi-mediated vit-1/2 knockdown [12]. This careful approach enabled the researchers to confidently conclude that vit-2 functions downstream of pry-1 to regulate lipid levels and lifespan, a finding that relied on precise measurement of transcript levels.
Table 2: Research Reagent Solutions for qRT-PCR Validation
| Reagent / Material | Function in Experiment | Example from Vitellogenin Study |
|---|---|---|
| Sequence-Specific Primers | Amplify the target cDNA for detection and quantification. | vit-2 primers (FP: GACACCGAGCTCATCCGCCCA, RP: TTCCTTCTCTCCATTGACCT) [12]. |
| Double-Quenched Probes | Report target amplification via fluorescence; double-quenching reduces background. | Recommended for TaqMan assays for higher signal-to-noise ratio [60]. |
| SYBR Green Master Mix | Intercalating dye that fluoresces when bound to double-stranded DNA. | SensiFAST SYBR Green Kit was used for qRT-PCR [12]. |
| SensiFAST cDNA Kit | Efficiently converts RNA to cDNA for the PCR template. | Used for reverse transcription with oligo(dT) primers [12]. |
| RNA Stabilization Solution | Preserves RNA integrity in fresh or stored tissue samples. | Products like RNAlater prevent degradation, which is critical for accuracy [58]. |
| DNA Decontamination Solution | Destroys contaminating DNA on surfaces to prevent false positives. | Use of DNAzap before setting up reactions for NAC control [58]. |
| Validated Reference Gene | Normalizes sample-to-sample variation in RNA quantity and quality. | pmp-3 was used as a stable reference gene for C. elegans studies [12]. |
Optimal primer and probe design is a critical, non-negotiable step in generating publication-quality qRT-PCR data, especially in complex experiments like vitellogenin knockdown validation. By adhering to strict thermodynamic parameters, rigorously screening for secondary structures using sophisticated tools, and employing comprehensive experimental validation, researchers can completely avoid the pitfalls of dimers and non-specific amplification. The comparative data and protocols provided here serve as a guide for objectively selecting design strategies that ensure the highest levels of accuracy and reproducibility, thereby solidifying the foundation upon which reliable biological conclusions are built.
In quantitative real-time PCR (qRT-PCR) validation of gene knockdown experiments, such as those targeting the vitellogenin (Vg) gene, precision is paramount. The reliability of your results depends significantly on two fundamental reaction components: primer concentration and master mix selection. Proper optimization of these elements ensures accurate quantification of transcript reduction, confirming the efficacy of RNA interference (RNAi) treatments. This guide provides an objective comparison of optimization strategies and products, supported by experimental data, to help researchers achieve robust and reproducible qRT-PCR results in their functional genomics research.
Primer Concentration Optimization
The Role of Primer Concentration
Primer concentration directly influences the specificity, efficiency, and sensitivity of qRT-PCR assays. Insufficient primer concentration can lead to poor sensitivity and low amplification efficiency, while excessive concentration promotes non-specific amplification and primer-dimer formation [65]. Optimal concentration is therefore critical for accurately measuring changes in gene expression, such as the knockdown of a target gene like vitellogenin.
General Recommendations and Polymerase-Specific Guidelines
The ideal primer concentration varies depending on the DNA polymerase used in the master mix. Archaeal Family B polymerases (e.g., Q5, Phusion) possess strong 3´-5´ exonuclease activity, which can digest nucleotides on the 3' end of primers. To counteract this effect and promote specific product formation, a higher final primer concentration of 500 nM is recommended [66]. In contrast, for Taq-based polymerases (e.g., OneTaq, standard Taq), a final concentration of 200 nM is typically sufficient [66].
The table below summarizes the manufacturer-recommended final primer concentrations for various common polymerases.
Table 1: Manufacturer-Recommended Primer Concentrations for Different Polymerases
| Polymerase | Recommended Final Concentration (each primer) | Final Concentration Range (each primer) |
|---|---|---|
| Q5 Polymerases | 500 nM | 200-1000 nM |
| Phusion Polymerases | 500 nM | 200-1000 nM |
| OneTaq Polymerases | 200 nM | 50-1000 nM |
| Taq Polymerases | 200 nM | 50-1000 nM |
| Hemo KlenTaq | 300 nM | 50-1000 nM |
| LongAmp Polymerases | 400 nM | 50-1000 nM |
Experimental Optimization Protocol
Relying solely on general guidelines is insufficient for a rigorously optimized assay. A systematic experimental approach is required.
- Test a Concentration Series: Prepare a dilution series of your primers, typically ranging from 50 nM to 900 nM, and test them in qPCR reactions using a fixed amount of template [67].
- Assess Specificity: Analyze the results using melt curve analysis (for SYBR Green assays). A single, sharp peak indicates specific amplification, while multiple peaks suggest primer-dimer formation or non-specific products that require further optimization [65].
- Evaluate Efficiency: Use a template serial dilution (at least 5 dilution steps) to generate a standard curve. The optimal primer concentration should yield a PCR efficiency between 90–110% and a linearity (R²) ≥ 0.99 [67]. The goal is to achieve 100 ± 5% efficiency, which is a prerequisite for using the 2−ΔΔCt method for data analysis [68].
Master Mix Selection and Optimization
Types of Master Mixes
The choice of master mix dictates the chemistry and overall performance of your qRT-PCR assay. The two most common approaches are:
- SYBR Green-Based Master Mixes: These utilize intercalating dyes that bind to double-stranded DNA, providing a fluorescent signal proportional to the amount of PCR product. They are more cost-effective and flexible but require careful primer design and post-run melt curve analysis to ensure specificity [65].
- TaqMan Probe-Based Master Mixes: These use sequence-specific hydrolysis probes labeled with a fluorophore and quencher. They offer higher specificity, are ideal for multiplexing, and do not require melt curve analysis, but are more expensive [65] [69]. A properly optimized SYBR Green assay can be as effective as a TaqMan assay for gene expression analysis [65].
Comparative Performance Data
The selection of a master mix can significantly impact key assay parameters like the Limit of Detection (LOD) and PCR efficiency. A 2021 comparative study evaluated seven commercial TaqMan master mixes for the detection of porcine DNA, providing objective performance data [70].
Table 2: Performance Comparison of Seven Commercial TaqMan Master Mixes [70]
| Master Mix (Manufacturer) | Reported LOD (pg/reaction) | Reported PCR Efficiency (%) |
|---|---|---|
| PowerAmp Real-time PCR Master Mix II (Kogene Biotech) | 0.5 pg | Not Specified |
| Express qPCR Supermix Universal (Invitrogen) | 0.5 pg | Not Specified |
| QuantiNova Probe PCR Kit (Qiagen) | 0.5 pg | Not Specified |
| Luna Universal Probe qPCR Master Mix (New England Biolabs) | 0.5 pg | Not Specified |
| TaqMan Universal PCR Master Mix (Applied Biosystems) | 0.5 - 5 pg* | Not Specified |
| MG 2X qPCR MasterMix (CancerROP) | 0.5 - 5 pg* | Not Specified |
| Premix Ex Taq (Takara) | 5 pg | Not Specified |
Note: LOD for these mixes varied depending on the real-time PCR platform used.
The study concluded that the sensitivity and specificity of a real-time PCR assay can vary significantly depending on the master mix and platform used, highlighting the importance of this selection [70].
Master Mix Optimization Checklist
Beyond selecting a mix, ensure the following parameters are optimized for your specific reaction:
- Passive Reference Dye (ROX): Use the correct ROX concentration (High, Low, or No ROX) as required by your qPCR instrument to normalize fluorescence signals across the plate [65] [67].
- Template Quality and Quantity: Use high-quality, purified RNA. For total RNA, a standard input range of 100 ng–10 pg is often recommended. Treat samples with DNase I to minimize genomic DNA amplification [67].
- Amplicon Design: Keep amplicons short, between 70–200 bp, for maximum PCR efficiency. The target sequence should have a GC content of 40–60% and avoid secondary structures [67].
- Thermal Cycling Conditions: Follow the manufacturer's recommended cycling conditions. A common default is 40 cycles, but this may be extended to 45 for very low-input samples [67].
Application in Vitellogenin Knockdown Research
In a 2021 study on silencing the vitellogenin (Vg) gene in the red palm weevil (Rhynchophorus ferrugineus), qRT-PCR was crucial for validating knockdown efficacy [71]. The researchers observed that suppressing RfVg expressions by 95-99% resulted in a dramatic failure of Vg protein expression, causing atrophied ovaries and a halt in egg hatchability [71]. Such definitive conclusions rely entirely on a qRT-PCR assay that has been finely tuned through proper optimization of primer concentrations and master mix components to avoid both false-positive and false-negative results.
The Scientist's Toolkit: Essential Reagents for qRT-PCR Optimization
Table 3: Key Reagents for qRT-PCR Optimization and Their Functions
| Reagent / Kit | Primary Function |
|---|---|
| High-Fidelity DNA Polymerase (e.g., Q5, Phusion) | Amplification of template DNA with high accuracy, often requiring higher primer concentrations [66]. |
| Taq DNA Polymerase | Standard polymerase for routine qPCR; used with lower primer concentrations [66]. |
| SYBR Green Master Mix | Provides intercalating dye for detection of double-stranded DNA amplification in a cost-effective manner [65]. |
| TaqMan Probe Master Mix | Provides chemistry for sequence-specific hydrolysis probes for highly specific target detection [65]. |
| DNase I (RNase-free) | Digests contaminating genomic DNA in RNA samples prior to reverse transcription [67]. |
| dNTPs | Building blocks (nucleotides) for DNA synthesis during PCR amplification. |
| Nuclease-Free Water | Solvent for diluting primers and templates, free of RNases and DNases that would degrade the reaction. |
Workflow and Pathway Diagrams
qPCR Optimization Workflow
The following diagram illustrates a systematic workflow for optimizing primer concentration and master mix in a qRT-PCR assay.
Vitellogenin Research Pathway
This diagram outlines the logical pathway connecting qRT-PCR optimization to functional conclusions in vitellogenin knockdown research.
The journey to reliable qRT-PCR data, especially for critical validations like gene knockdown, is built on the meticulous optimization of reaction components. There is no universal "best" primer concentration or master mix; the optimal combination is determined empirically for each assay system. By systematically testing primer concentrations against the backdrop of a carefully selected master mix, researchers can achieve the high efficiency and specificity required to draw confident biological conclusions. The data and protocols provided here serve as a guide for researchers to fine-tune their reactions, ensuring that their findings, such as the successful knockdown of vitellogenin, are beyond reproach.
The reliability of qRT-PCR data in vitellogenin (Vg) knockdown research is fundamentally dependent on the meticulous control of pre-analytical variables. Vg, a conserved glycolipoprotein, is central to egg-yolk formation but also exhibits roles in immunity, antioxidant activity, and, as recent evidence in honey bees suggests, potential involvement in gene regulation through DNA-binding activity [3]. The quantification of Vg mRNA levels following experimental knockdown requires precise and reproducible methods to avoid artifacts that could compromise data interpretation. This guide objectively compares methodologies and solutions for sample collection, RNA integrity assessment, and cDNA synthesis, providing a standardized framework to enhance the validity of research findings in this field.
Sample Collection and Stabilization
The pre-analytical phase begins immediately upon sample collection, where RNA integrity is most vulnerable. Proper handling and stabilization are critical for preserving the accurate in vivo mRNA profile, including transcripts for Vg.
Table 1: Comparison of Blood Collection and Sample Storage Methods for RNA Analysis
| Method / Condition | Key Features | Impact on RNA Integrity | Best Use Cases |
|---|---|---|---|
| K₂EDTA Tubes [72] | - Anticoagulant- Requires rapid processing (<2-6 hrs at RT) [72]- Ex vivo gene expression changes can start immediately [72] | High risk of RNA degradation and gene expression changes if processing is delayed. | Research settings where immediate processing is feasible. |
| PAXgene Blood RNA Tubes [72] | - Contains RNA-stabilizing additives | Effectively stabilizes RNA, minimizing degradation and ex vivo changes. | Clinical studies, biobanking, and multi-site trials. |
| RNAlater / Similar Solutions [73] | - Aqueous, non-toxic solution- Inactivates RNases by precipating cellular proteins- Permits storage at 4°C or -20°C after immersion | Excellent for tissue samples, preserving RNA integrity outside a freezer. | Field collections, solid tissues (e.g., liver, fat body). |
| Snap-Freezing (Fresh Frozen) [73] | - Immediate immersion in liquid nitrogen- Requires continuous storage at -80°C | The gold standard for tissue preservation when performed correctly. | Most laboratory settings where liquid nitrogen is available. |
| Room Temperature Storage [73] | - No stabilization- Highly variable and sample-dependent | Rapid degradation; not recommended for RNA work. | Avoid for RNA analysis. |
Experimental Protocol: Assessing Sample Stability. To validate a sample collection workflow, a time-course experiment can be performed. As demonstrated in a study identifying RNA quality biomarkers, blood collected in K₂EDTA and PAXgene tubes should be stored at different temperatures (e.g., 4°C and room temperature) and for varying durations (e.g., 0, 2, 6, 24, 48, and 72 hours) before RNA extraction [72]. The resulting RNA can then be analyzed using methods described in Section 2 to determine the acceptable storage window for specific sample types.
RNA Quality Assessment Methods
Once a sample is stabilized, assessing RNA quality is a mandatory step before proceeding to cDNA synthesis. The following methods provide complementary data on RNA quantity, purity, and integrity.
Table 2: Comparison of RNA Quality Control Methods
| Method | Principle | Information Provided | Advantages | Limitations |
|---|---|---|---|---|
| UV Spectrophotometry (e.g., NanoDrop) [74] [75] | Absorbance of UV light by nucleic acids at 260 nm. | - Concentration (A260)- Purity (A260/A280 ~2.0 for pure RNA; A260/A230 >1.8) [75] | Fast, requires minimal sample volume (1-2 µl), non-destructive. | Cannot distinguish between RNA and DNA; insensitive to degradation; overestimates concentration if contaminants are present. |
| Fluorometry (e.g., Qubit with RNA-specific dyes) [74] [75] | Fluorescence emission from dyes binding specifically to RNA. | - Accurate RNA concentration- High sensitivity | Highly specific and sensitive for RNA; accurate for low-concentration samples. | Requires specific dyes and equipment; does not assess integrity or purity from contaminants like salts. |
| Denaturing Agarose Gel Electrophoresis [76] [74] | Separation of RNA by size in a gel matrix. | - Integrity: Sharp 28S and 18S rRNA bands with a 2:1 intensity ratio indicate intact RNA [76]. | Low cost; provides visual evidence of degradation (smearing) and gDNA contamination (high molecular weight band). | Semi-quantitative; requires hundreds of nanograms of RNA; uses hazardous stains (EtBr); less sensitive. |
| Automated Capillary Electrophoresis (e.g., Agilent Bioanalyzer) [72] [74] [75] | Microfluidics-based separation and fluorescence detection. | - Concentration- Integrity: RNA Integrity Number (RIN) on a 1 (degraded) to 10 (intact) scale.- Size distribution | Highly reproducible; requires very little sample (1 µl); provides a numerical integrity score (RIN/RQI) for objective comparison. | Higher cost per sample; requires specialized equipment and chips. |
| 3'/5' or Short/Medium/Long Assays [72] [73] | qPCR-based assays comparing amplification from different regions of a transcript. | - mRNA integrity: A difference in Cq values indicates targeted mRNA degradation. | Assesses the integrity of the actual mRNA target, not just rRNA; highly sensitive. | Requires validated qPCR assays; does not provide a holistic view of total RNA quality. |
Experimental Protocol: The 3'/5' Integrity Assay. This method is crucial for validating RNA quality for RT-qPCR, as ribosomal RNA integrity does not always reflect mRNA quality [72] [73]. For a target gene like Vitellogenin, two qPCR assays are designed: one near the 3' end and one near the 5' end of the transcript. The difference in quantification cycle (ΔCq = Cq5' - Cq3') is calculated. A small ΔCq indicates intact mRNA. A threshold ratio (e.g., 3':5' ratio between 0.2 and 5) should be established to define acceptable sample quality [73]. It is critical that both assays have virtually identical PCR efficiencies for accurate comparison [73].
Diagram 1: RNA QC workflow. This chart outlines the pathway for comprehensive RNA quality assessment, from initial sample to final decision.
cDNA Synthesis: Critical Steps and Optimization
The reverse transcription reaction is a major source of variability in RT-qPCR. Optimizing this step is vital for accurate quantification of Vg mRNA.
Table 3: Comparison of Reverse Transcriptase Enzymes for cDNA Synthesis
| Attribute | AMV Reverse Transcriptase [77] | MMLV Reverse Transcriptase [77] | Engineered MMLV (e.g., SuperScript IV) [77] |
|---|---|---|---|
| RNase H Activity | High | Medium | Low / None (RNaseH-) |
| Reaction Temperature | Up to 42°C | Up to 37°C | Up to 55°C |
| Reaction Time | ~60 minutes | ~60 minutes | ~10 minutes |
| cDNA Yield (with challenging RNA) | Medium | Low | High |
| Ideal for | Standard templates with low secondary structure. | Routine cDNA synthesis. | GC-rich templates, RNAs with secondary structure, long transcripts. |
Experimental Protocol: Efficient cDNA Synthesis.
- Genomic DNA Removal: Treat purified RNA with a DNase to eliminate gDNA contamination. Thermolabile DNases are preferred as they can be inactivated by a brief heating step (e.g., 5 min at 55°C), preventing RNA loss from purification procedures [77].
- Primer Selection:
- Oligo(dT) primers: Best for amplifying mRNAs with poly-A tails, providing strand-specific cDNA. Ideal for Vg mRNA.
- Random hexamers: Prime throughout the RNA population, including degraded RNA and non-polyadenylated RNAs.
- Gene-specific primers: Most specific but only suitable for one target per reaction.
- Reverse Transcription Reaction:
- Prepare the reaction mix containing RNA template, reaction buffer, dNTPs (0.5-1 mM each), DTT, RNase inhibitor, selected primers, and reverse transcriptase [77].
- For RNAs with high GC-content or secondary structure, include an optional denaturation step: heat RNA with primers to 65°C for 5 minutes, then immediately chill on ice [77].
- Perform the synthesis using the temperature and duration optimal for the chosen enzyme (see Table 3). Engineered MMLV enzymes allow higher reaction temperatures (50-55°C), which help denature secondary structures, resulting in higher cDNA yield and better representation of the transcriptome [77].
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table lists key reagents and their critical functions in the pre-analytical workflow for Vg research.
Table 4: Essential Reagents for Pre-analytical Workflow in Gene Expression Studies
| Reagent / Solution | Function in Workflow |
|---|---|
| RNAlater or Similar RNA Stabilization Reagent [73] | Preserves RNA integrity in tissues and cells immediately after collection, inactivating RNases. |
| PAXgene Blood RNA Tubes [72] | Provides immediate stabilization of RNA in whole blood, critical for clinical Vg studies. |
| Acidic-Phenol Guanidinium Thiocyanate-based Lysis Buffers (e.g., TRIzol) | Effective simultaneous lysis and denaturation of nucleases during RNA extraction from challenging samples. |
| DNase I (or thermolabile DNase) [77] | Digests contaminating genomic DNA in RNA preparations to prevent false-positive signals in qPCR. |
| RNase Inhibitors [77] | Added to reverse transcription reactions to protect RNA templates from degradation by RNases. |
| Engineered MMLV Reverse Transcriptase (RNase H-) [77] | High-efficiency enzyme for synthesizing high-yield, full-length cDNA, even from suboptimal RNA. |
Impact on qRT-PCR Data Analysis and Vg Validation
The choices made in the pre-analytical phase directly impact the qRT-PCR results. The quantification cycle (Cq) is not only dependent on the initial target concentration but also on PCR efficiency and the setting of the quantification threshold [78]. Ignoring these factors can lead to severe miscalculations. For example, interpreting a ΔCq value without correcting for PCR efficiency can lead to an assumed gene expression ratio that is 100-fold off from the true value [78]. Furthermore, the use of degraded RNA can lead to an underestimation of transcript levels, which is particularly dangerous in knockdown studies where it could exaggerate the apparent knockdown efficiency [72] [73]. Validating RNA integrity with methods like the 3'/5' assay and using efficiency-corrected calculations for relative quantification are therefore non-negotiable for robust Vg validation.
Diagram 2: Pre-analytical errors impact. This diagram maps the relationship between common pre-analytical errors and their downstream effects on qRT-PCR data quality.
The path to reliable Vg knockdown validation via qRT-PCR is paved long before the PCR plate is loaded. It begins at the moment of sample collection and is cemented by rigorous quality control of RNA and an optimized cDNA synthesis protocol. By systematically addressing pre-analytical variables through the methodologies and comparisons outlined in this guide, researchers can significantly reduce technical noise, enhance the reproducibility of their experiments, and draw biologically meaningful conclusions about the complex functions of vitellogenin.
Inconsistent gene knockdown remains a significant hurdle in RNA interference (RNAi) research, particularly in the context of qRT-PCR validation of vitellogenin knockdown. The efficacy of RNAi is fundamentally governed by two interconnected factors: the inherent stability of the double-stranded RNA (dsRNA) trigger and the efficiency of its delivery into the target cells. This guide provides a comparative analysis of current technologies and methodologies, presenting objective experimental data to help researchers troubleshoot and optimize their silencing experiments, ensuring reliable and reproducible results.
The Core Challenge: dsRNA Instability and Degradation
A primary cause of inconsistent knockdown is the rapid degradation of dsRNA before it reaches its target. Specific nucleases, known as dsRNases, are major contributors to this problem, and their expression varies significantly across species and tissues.
- Evidence from Lepidopteran Pests: Research on Spodoptera exigua identified four dsRNase genes (SeRNase1-4). Expression analysis revealed that these genes are highly active in the midgut, a key interface for oral RNAi, creating a hostile environment for naked dsRNA [79].
- Impact on RNAi Efficacy: A study on Spodoptera litura demonstrated that despite its relative environmental stability, dsRNA undergoes rapid degradation within the larval gut. Northern blot analyses confirmed the inability to recover intact dsRNA or detect functional siRNA processing following oral delivery, directly linking nuclease activity to RNAi failure [80].
The following table summarizes the differential RNAi efficacy observed in a direct comparison study.
Table 1: Comparative Efficacy of dsRNA vs. siRNA in Spodoptera litura [80]
| RNAi Trigger | Target Gene | Mortality Effect | Gene Silencing Efficiency | Key Finding |
|---|---|---|---|---|
| Long dsRNA | mesh |
Not Significant | Low | Failed to produce functional siRNAs in the midgut. |
| Long dsRNA | iap |
Not Significant | Low | Rapid degradation in the gut environment. |
| siRNA | mesh |
Significant | High | Effective gene silencing and larval mortality. |
| siRNA | iap |
Significant | High | Bypasses the need for Dicer-2 processing. |
Advanced RNA Formulations to Enhance Stability
To overcome instability, advanced RNA formulations that protect the payload from nucleases have been developed. These platforms represent a significant leap beyond traditional dsRNA.
- Self-Assembled RNA Nanostructures (SARNs): This technology involves engineering single-stranded RNAs that self-fold into defined nanostructures, capable of loading pools of siRNA duplexes. Key advantages include:
- Enhanced Stability: SARNs exhibit superior resistance to environmental nucleases compared to traditional dsRNA [81] [82].
- Programmable Release: They are designed with motifs that enable both immediate and sustained release of siRNAs, leading to more durable silencing [81].
- Improved Cellular Uptake: The nanostructures have favorable hydrophobicity and elasticity, enhancing their ability to enter cells [81].
- Efficacy Data: In models with both chewing (Tribolium castaneum) and piercing-sucking (Nilaparvata lugens) mouthparts, SARNs demonstrated significantly higher RNAi efficiency and mortality compared to conventional dsRNA [81] [82].
Table 2: Performance of Engineered SARNs vs. Traditional dsRNA [81] [82]
| Feature | Traditional dsRNA | Engineered SARNs |
|---|---|---|
| Nuclease Resistance | Low | High |
| Cellular Uptake | Variable and often inefficient | Enhanced (hydrophobicity/elasticity) |
| Silencing Kinetics | Single-phase, short-lived | Immediate + sustained release |
| Production Scalability | Challenging for some formats | Scalable via E. coli transcription |
| Efficacy in Piercing-Sucking Pests | Generally low | Demonstrated high efficacy in Nilaparvata lugens |
Delivery System Technologies
The choice of delivery system is critical for shielding dsRNA during transit and facilitating its cellular internalization.
- Nanoparticle-Mediated Delivery: The instability posed by dsRNases in Spodoptera exigua was successfully mitigated using a star polycation (SPc) nanocarrier [79]. This complex protects dsRNA by binding it and preventing SeRNase access. The system facilitates clathrin-mediated endocytosis and promotes endosomal escape, ensuring intracellular dsRNA release [79].
- Non-Cationic Carriers: While cationic lipids/polymers are common, their cytotoxicity can be a limitation. Non-cationic carriers (e.g., certain organic polymers, inorganic materials, and biomimetic vectors) offer superior biocompatibility. siRNA loading is achieved through strategies like chemical conjugation, hydrogen bonding, and hydrophobic interactions, which are highly relevant for in vivo applications [83].
- Comparative Delivery Methods in Mosquitoes: A study on Aedes albopictus larvae compared three non-invasive delivery methods—soaking, rehydration, and nanoparticle ingestion. The results were gene-dependent, but rehydration was the most specific and efficient overall. This highlights the need for empirical optimization of delivery routes for specific experimental systems [84].
The Scientist's Toolkit: Essential Research Reagents
The following reagents and kits are critical for conducting and validating RNAi experiments, as evidenced by the protocols in the surveyed literature.
Table 3: Key Research Reagent Solutions for RNAi Experiments
| Reagent / Kit Name | Function in Experiment | Example Use Case |
|---|---|---|
| TRIzol Reagent | Total RNA isolation from tissues and cells. | RNA extraction from insect midguts for qRT-PCR [80]. |
| MEGAscript T7 Kit | In vitro transcription for high-yield dsRNA synthesis. | Production of target-specific dsRNA for feeding assays [80]. |
| mirVana miRNA Isolation Kit | Purification of small RNA fractions (<200 nt). | Enrichment of siRNAs for northern blot analysis [80]. |
| SensiFAST SYBR Hi-ROX Kit | One-step SYBR Green-based mix for qRT-PCR. | Quantitative validation of gene knockdown (e.g., vitellogenin mRNA levels) [80]. |
| PrimeScript RT Reagent Kit | High-efficiency reverse transcription of RNA to cDNA. | First-strand cDNA synthesis prior to qPCR amplification [80]. |
| ZR small-RNA PAGE Recovery Kit | Purification and recovery of small RNAs from polyacrylamide gels. | Isolation of specific siRNA bands following northern blot electrophoresis [81]. |
Experimental Workflow for qRT-PCR Validation of Knockdown
The diagram below outlines a robust experimental workflow, from design to validation, integrating strategies to overcome stability and delivery issues.
The RNAi Pathway and Key Barriers
A molecular-level understanding of the RNAi pathway helps identify where failures can occur. The following diagram maps the journey of an exogenous dsRNA trigger to gene silencing, highlighting critical barriers like nuclease degradation and inefficient Dicing.
Resolving inconsistent knockdown in RNAi experiments requires a holistic approach that addresses both the biochemical stability of the RNA trigger and the biophysical challenge of delivering it intracellularly. As the data demonstrates, moving beyond naked dsRNA to advanced solutions like SARNs, siRNA, and nanoparticle encapsulation can dramatically improve nuclease resistance and uptake. The choice of delivery method must be empirically optimized for the specific biological system. By systematically applying these strategies and employing rigorous qRT-PCR validation protocols, researchers can achieve the reliable and potent gene silencing necessary for robust scientific conclusions, including in complex studies such as vitellogenin knockdown.
In vitellogenin (Vg) knockdown research, the reliability of qRT-PCR data is paramount for drawing accurate conclusions about gene function. The implementation of robust experimental controls is a critical defense against artifacts and false results. These controls validate every step of the process, from RNA extraction to final amplification, ensuring that observed expression changes genuinely reflect experimental manipulation rather than technical inconsistencies. Within the context of Vg research—where this highly conserved protein influences diverse biological processes from reproduction to antioxidant defense—precise quantification is essential for elucidating its multifunctional roles [3].
This guide objectively compares control implementations and provides standardized protocols for their application in Vg knockdown studies. We present experimental data and structured methodologies to equip researchers with the tools necessary for implementing rigorous qRT-PCR controls, thereby enhancing data credibility and reproducibility in gene expression analysis.
The Control Ecosystem: Purposes and Implementation
Defining the Control Hierarchy
Table 1: Core qRT-PCR Controls in Gene Knockdown Experiments
| Control Type | Primary Purpose | Implementation Method | Interpretation of Results |
|---|---|---|---|
| No-Template Control (NTC) | Detects reagent contamination or primer-dimer formation [85]. | Contains all reaction components except template DNA/cRNA, replaced with nuclease-free water. | A negative result (no amplification) validates reagent purity. A positive result (amplification) indicates contamination requiring investigation [85]. |
| Extraction Control | Monitors efficiency and potential contamination during nucleic acid isolation. | Incorporates the same biological matrix as samples through identical extraction process. | Successful amplification confirms effective nucleic acid recovery. Unexpected amplification signals potential cross-contamination. |
| Amplification Control | Assesses PCR inhibition and reaction efficiency. | Uses a known quantity of exogenous control template or validated reference genes. | Amplification within expected parameters indicates unimpeded reaction kinetics. Shifted Cq values suggest PCR inhibitors. |
| Reverse Transcription Control | Verifies cDNA synthesis efficiency and gDNA contamination. | Includes reactions without reverse transcriptase enzyme (-RT control). | Amplification in -RT control indicates genomic DNA contamination, necessitating DNase treatment or improved primer design. |
Consequences of Inadequate Controls
Failure to implement appropriate controls can compromise experimental conclusions. For instance, amplification in NTCs can result from several issues:
- Reagent contamination occurs when one or more reaction components are contaminated, typically producing consistent amplification across NTC replicates [85].
- Primer-dimer formation, common with SYBR Green chemistry, generates nonspecific amplification that increases background noise and may yield Cq values <40, potentially altering expression interpretation for experimental samples [85].
- Carryover contamination from previous PCR products can be mitigated through physical separation of pre- and post-PCR workspaces and incorporating uracil-N-glycosylase (UNG) into master mixes [85].
In SARS-CoV-2 diagnostics, unspecific amplification in negative samples and NTCs occurred in 56.4% and 57.1% of cases, respectively, due to dimer formation in primer-probe sets. Optimizing RT-qPCR parameters reduced these false signals to 11.5%, highlighting the critical importance of validation [86].
Experimental Protocols for Control Implementation
Sample Preparation and RNA Extraction
Protocol: RNA Extraction with Controlled Workflow
- Tissue Collection: Dissect target tissues (e.g., fat body, hypopharyngeal glands, brain) under a microscope in honeybees [87]. Immediately freeze samples in liquid nitrogen.
- Homogenization: Grind tissues in TRIzol reagent using polyvinylpyrrolidone (PVP) to remove polysaccharides and polyphenols [88].
- RNA Extraction: Use commercial kits (e.g., TransZol Up Plus RNA Kit, TIANGEN RNAprep Plant Kit) following manufacturer protocols [89] [88].
- DNA Removal: Treat all RNA samples with RNase-free DNase I to minimize gDNA contamination [88].
- Quality Assessment: Verify RNA integrity via 1.2% agarose gel electrophoresis and determine concentration/purity using spectrophotometry (NanoDrop) [87] [88].
- Extraction Control: Process a mock sample (nuclease-free water plus biological matrix) alongside experimental samples.
cDNA Synthesis and Control Setup
Protocol: cDNA Synthesis with RT Controls
- Reverse Transcription: Synthesize cDNA using kits (e.g., PrimeScript RT reagent Kit, EasyScript One-Step gDNA Removal cDNA Synthesis SuperMix) with 1μg RNA input [87] [89].
- -RT Control Preparation: For each sample, include a reaction without reverse transcriptase enzyme.
- Storage: Store cDNA at -20°C for long-term preservation [88].
qRT-PCR Setup and Control Reactions
Protocol: qPCR Plate Setup with Essential Controls
- Reaction Preparation: Use commercial premixes (e.g., TB Green Premix Ex Taq II, 2× SuperReal PreMix Plus) in 20μL reactions [87] [88].
- Template Addition: Add 2μL of diluted (1:5) cDNA per reaction [88].
- Control Reactions:
- NTC: Replace template with nuclease-free water.
- Extraction Control: Use nucleic acid from the extraction control.
- Positive Amplification Control: Use a validated reference gene or synthetic template.
- Thermocycling Conditions:
- Melting Curve Analysis: Perform dissociation protocol following amplification when using SYBR Green chemistry to identify primer-dimer formation [85] [88].
Data Analysis and Validation
Troubleshooting Control Failures
Table 2: Troubleshooting Guide for Control Anomalies
| Control Failure | Potential Causes | Corrective Actions |
|---|---|---|
| NTC Amplification | - Contaminated reagents [85]- Primer-dimer formation [85]- Carryover contamination [85] | - Prepare fresh reagents- Optimize primer concentrations [85]- Implement UNG treatment [85]- Use separate work areas [85] |
| Extraction Control Amplification | - Cross-contamination during extraction- Contaminated extraction reagents | - Decontaminate work surfaces- Use fresh aliquots of extraction reagents- Implement dedicated pipettes |
| Irregular Cq in Positive Control | - PCR inhibitors- Reagent degradation- Pipetting errors | - Purify template DNA- Prepare fresh master mix- Calibrate pipettes- Include dilution series for efficiency calculation |
| Amplification in -RT Control | - Genomic DNA contamination- Inefficient DNase treatment | - Optimize DNase treatment protocol- Design primers spanning exon-exon junctions |
Validation Against Reference Genes
In vitellogenin research, particularly in honeybees, proper validation includes using stable reference genes. Studies have identified ADP-ribosylation factor 1 (arf1) and ribosomal protein L32 (rpL32) as the most stable reference genes across honeybee tissues and developmental stages, while conventional genes like α-tubulin, glyceraldehyde-3-phosphate dehydrogenase (gapdh), and β-actin displayed poor stability [87]. This emphasizes the necessity of validating reference genes rather than assuming their stability.
Figure 1: Integrated Control Workflow for qRT-PCR Experiments. This diagram illustrates the sequential implementation of essential controls throughout the qRT-PCR process, from RNA extraction to data analysis.
Research Reagent Solutions
Table 3: Essential Reagents for qRT-PCR Control Implementation
| Reagent/Category | Specific Examples | Function in Control Implementation |
|---|---|---|
| RNA Extraction Kits | TRIzol reagent (Invitrogen) [87], TransZol Up Plus RNA Kit (TransGen) [89], TIANGEN RNAprep Plant Kit [88] | Isolate high-quality RNA while monitoring extraction efficiency and potential contamination. |
| DNA Removal Reagents | RNase-free DNase I [88], gDNA wipe buffer (TIANGEN FastQuant RT Kit) [88] | Eliminate genomic DNA contamination, critical for validating -RT controls. |
| Reverse Transcription Kits | PrimeScript RT reagent Kit (TaKaRa) [87], EasyScript cDNA Synthesis SuperMix (TransGen) [89] | Generate high-quality cDNA while enabling -RT control preparation. |
| qPCR Master Mixes | TB Green Premix Ex Taq II (TaKaRa) [87], 2× SuperReal PreMix Plus (TIANGEN) [88] | Provide optimized reaction components for consistent amplification across samples and controls. |
| Contamination Prevention | AmpErase UNG (Uracil-N-Glycosylase) [85], ROX Reference Dye [88] | Reduce carryover contamination between experiments and normalize fluorescence signals. |
The implementation of extraction, amplification, and no-template controls represents a non-negotiable standard in vitellogenin knockdown research using qRT-PCR. These controls provide the necessary framework for distinguishing technical artifacts from genuine biological signals, thereby ensuring data integrity. Through systematic implementation of the protocols and troubleshooting strategies outlined here, researchers can significantly enhance the reliability of their gene expression findings in Vg studies and beyond.
Validating Knockdown Impact: From Analytical Rigor to Phenotypic Confirmation
Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR) is a cornerstone technique in molecular biology for the sensitive and specific quantification of RNA expression levels. In functional genomics research, such as studies investigating vitellogenin (Vg) knockdown, robust analytical validation of the qRT-PCR method is paramount. This process ensures that observed changes in gene expression are accurate, reproducible, and biologically meaningful. Establishing method sensitivity, specificity, and precision provides the rigorous foundation required for reliable data interpretation in drug development and basic research [90]. This guide objectively compares the performance of qRT-PCR with emerging alternative technologies, namely digital PCR (dPCR), providing a framework for researchers to select the optimal tool for gene expression validation.
Core Analytical Performance Parameters
The validation of any qRT-PCR assay rests on three fundamental pillars: sensitivity, specificity, and precision. Each parameter addresses a distinct aspect of assay performance and must be empirically demonstrated.
- Sensitivity refers to the lowest quantity of the target nucleic acid that can be reliably measured. It is typically defined by the Limit of Detection (LoD), the lowest concentration at which the target can be detected in at least 95% of replicates, and the Limit of Quantification (LoQ), the lowest concentration that can be measured with acceptable precision and accuracy [91] [92]. In practice, qRT-PCR assays targeting vitellogenin must be sensitive enough to detect potentially small but biologically significant changes in mRNA levels following knockdown.
- Specificity is the ability of the assay to detect only the intended target sequence, in this case,
VgmRNA, without cross-reacting with non-target sequences, such as homologous genes or genomic DNA. This is often ensured through careful primer and probe design and confirmed using melt curve analysis (for dye-based assays) or the use of target-specific fluorescent probes [93] [90]. - Precision describes the reproducibility of the measurements, quantified as the degree of scatter between repeated analyses of the same sample. It is usually reported as the Coefficient of Variation (CV%) for intra-assay (within-run) and inter-assay (between-run) replicates. A validated assay must demonstrate low variability to ensure that differences in
Vgexpression are due to experimental intervention and not technical noise [26] [92].
qRT-PCR vs. dPCR: A Comparative Performance Analysis
While qRT-PCR remains the workhorse for gene expression analysis, digital PCR (dPCR) has emerged as a powerful alternative. The table below provides a structured comparison of their key performance characteristics, synthesizing data from multiple application studies.
Table 1: Performance comparison between qRT-PCR and dPCR
| Performance Parameter | qRT-PCR | dPCR | Experimental Context & Supporting Data |
|---|---|---|---|
| Sensitivity (LoD) | High (fg-pg of DNA) [26] | Very High; 10-100x lower LoD than qRT-PCR in some applications [94] | Probiotic Detection: ddPCR showed a 10- to 100-fold lower limit of detection compared to qRT-PCR for detecting bacterial strains in fecal samples [94]. |
| Absolute Quantification | No; requires a standard curve [91] | Yes; via Poisson statistics without a standard curve [91] [94] | Viral Detection: dPCR's absolute quantification is beneficial for measuring copy number variation and rare alleles without calibration curves [91]. |
| Precision (CV%) | Good to Excellent (e.g., CV <20%) [92] | Excellent; potentially higher precision, especially at low target concentrations [91] [94] | Residual DNA Testing: A validated qPCR assay for Vero cell DNA demonstrated high precision with a CV of less than 20% [92]. dPCR is noted for superior precision due to partitioning [91]. |
| Tolerance to PCR Inhibitors | Moderate | High; sample partitioning reduces inhibitor effects [91] [94] | Complex Matrices: dPCR is less susceptible to the effects of PCR inhibitors present in complex biological samples like feces, providing more robust detection [94]. |
| Throughput & Cost-Effectiveness | High; well-suited for high-throughput screening, lower cost per reaction [91] | Lower throughput; higher cost per reaction, especially for consumables [91] | Mass Screening: During the COVID-19 pandemic, qRT-PCR's high throughput and cost-effectiveness (as low as $0.2 per test) made it the predominant method for mass testing [91]. |
| Multiplexing Capability | Well-established for multiplexing with different fluorescent probes [90] | Challenging due to limited fluorescence channels and spectral overlap [91] | Variant Detection: qPCR melting curve assays allow for multiplexed detection of multiple SARS-CoV-2 mutations in a single reaction [93]. |
Experimental Protocols for Validation
Protocol for qRT-PCR Assay Validation
The following workflow outlines the key steps for validating a qRT-PCR assay, such as for detecting Vg mRNA expression changes.
Figure 1: A standard workflow for the development and validation of a qRT-PCR assay.
- Assay Design: Design primers and/or probes to target a unique region of the
Vgtranscript, distinguishing it from related genes. Bioinformatics analysis should confirm specificity [26] [3]. - RNA Extraction and Reverse Transcription: Extract high-quality total RNA from tissues of interest (e.g., fat body or hepatopancreas). Use a robust reverse transcription protocol to generate cDNA. The choice between one-step (combined RT and PCR) or two-step (separate reactions) protocols depends on throughput and the need to archive cDNA [90].
- Standard Curve and Validation Runs:
- Prepare a serial dilution of a known quantity of the target amplicon or a standardized cDNA sample to generate a standard curve.
- Run qPCR reactions in replicates (at least 3-5) across different days to assess inter-assay precision.
- Include no-template controls (NTCs) to check for contamination.
- Data Analysis:
- Linearity and Efficiency: From the standard curve, determine the correlation coefficient (R²) and amplification efficiency (ideally 90-110%) [92].
- Precision: Calculate the CV% for the Ct values of replicates at different concentrations.
- Sensitivity (LoD/LoQ): The LoD can be determined as the lowest concentration where 95% of positive replicates are detected. The LoQ is the lowest concentration with a CV% below an acceptable threshold (e.g., 25%) [92].
- Specificity: For dye-based assays, perform melt curve analysis to confirm a single, sharp peak at the expected melting temperature (Tm). For probe-based assays, sequence confirmation of the amplicon can be used [90].
Protocol for dPCR Assay
The fundamental steps for RNA extraction and cDNA synthesis are identical to qRT-PCR. The key differences lie in the preparation and reading of the PCR reaction.
- Sample Partitioning: The cDNA-containing PCR mix is partitioned into thousands to millions of individual nanoliter-sized droplets [94].
- Endpoint PCR: The droplet emulsion is cycled to endpoint on a conventional thermal cycler. Each droplet acts as an individual PCR reaction.
- Droplet Reading: The droplets are streamed past a fluorescent detector in a single file. Droplets are counted as positive (containing the target) or negative (not containing the target) based on their fluorescence amplitude [91] [94].
- Absolute Quantification: The absolute copy number concentration of the target in the original sample is calculated directly from the ratio of positive to negative droplets using Poisson statistics, eliminating the need for a standard curve [91].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key reagents and materials for qRT-PCR validation
| Item | Function in Experiment | Example from Literature |
|---|---|---|
| Sequence-Specific Primers/Probes | To amplify and detect the target Vg mRNA sequence with high specificity. |
Hydrolysis probes (TaqMan) or hairpin probes (Molecular Beacons) are used for specific detection, unlike non-specific DNA-binding dyes [93] [90]. |
| Reverse Transcriptase Enzyme | To synthesize complementary DNA (cDNA) from the RNA template. | A critical component for the first step in RT-qPCR, available in kits for 1-step or 2-step protocols [90]. |
| Hot-Start DNA Polymerase | To minimize non-specific amplification and primer-dimer formation during reaction setup. | A standard component of commercial qPCR master mixes to improve assay robustness and sensitivity [26] [93]. |
| Fluorescent Dyes or Labeled Probes | To generate the fluorescent signal proportional to the amount of amplified PCR product. | SYBR Green dye or probe-based systems like EasyBeacon probes are used for real-time detection [93] [92] [90]. |
| Nucleic Acid Extraction Kit | To purify high-quality, inhibitor-free RNA from complex biological samples. | Magnetic bead-based kits (e.g., MagMAX) are commonly used for automated, high-throughput nucleic acid extraction from tissues or cells [26] [94]. |
| Certified DNA/RNA Standards | To create a standard curve for qPCR quantification and for assessing linearity, efficiency, and LoD. | Studies use certified genomic DNA standards, such as Vero cell DNA, to generate calibration curves for validation [92]. |
Both qRT-PCR and dPCR are powerful technologies for the analytical validation of gene expression studies like vitellogenin knockdown. The choice between them is not a matter of which is universally superior, but which is most fit-for-purpose. qRT-PCR remains the ideal choice for high-throughput, cost-effective screening where relative quantification is sufficient and extreme sensitivity is not the primary concern. Its well-established protocols and multiplexing capabilities make it a versatile workhorse. In contrast, dPCR excels in applications requiring absolute quantification, exceptional sensitivity for rare targets, superior precision at low copy numbers, and robust performance in the presence of inhibitors. Researchers must weigh these performance characteristics, summarized in this guide, against their specific experimental needs, sample availability, and budgetary constraints to make an informed decision.
Vitellogenin (Vg), a phospholipoglycoprotein, is a critical yolk precursor protein in most oviparous organisms, providing the primary nutrient source for embryonic development [48] [4]. In insects, Vg is typically synthesized in the fat body, secreted into the hemolymph, and transported to the oocytes, where it is internalized by receptor-mediated endocytosis to form vitellin (Vt), the main yolk protein [4]. Beyond its fundamental role in reproduction, recent research has revealed that Vg and its homologs have undergone evolutionary co-option and pleiotropic expansion, influencing diverse physiological processes including behavioral maturation, task specialization, immune response, and lifespan regulation [2] [4] [14]. This guide synthesizes experimental data from RNA interference (RNAi) studies, primarily validated through quantitative reverse transcription polymerase chain reaction (qRT-PCR), to compare phenotypic outcomes resulting from Vg knockdown across multiple insect species. The correlation between molecular data—particularly gene expression levels—and the resulting phenotypes provides crucial insights for researchers investigating reproductive biology, behavioral ecology, and novel pest control strategies.
Experimental Approaches for Vitellogenin Functional Analysis
Core Methodological Framework: RNAi and qRT-PCR Validation
The standard experimental paradigm for investigating Vg function involves a coordinated approach combining gene silencing and molecular validation:
- RNAi-Mediated Knockdown: Double-stranded RNA (dsRNA) targeting Vg transcripts is introduced via injection or feeding, triggering sequence-specific degradation of complementary mRNA [95] [48] [2].
- qRT-PCR Validation: Successful knockdown is confirmed through qRT-PCR, which quantifies remaining transcript levels by reverse transcribing RNA to DNA, amplifying the gene of interest with specific primers, and measuring amplification using intercalating dyes (e.g., SYBR Green) or sequence-specific probes (e.g., Taqman) [96].
- Phenotypic Assessment: Validated knockdowns are systematically evaluated for reproductive, behavioral, and physiological outcomes through morphological examination, behavioral assays, and survivorship tracking [48] [2] [4].
Table 1: Key Experimental Components for Vitellogenin Functional Analysis
| Component | Function | Examples & Specifications |
|---|---|---|
| dsRNA Template | Triggers sequence-specific gene silencing | Target-specific sequence; gel verification recommended [96] |
| qRT-PCR System | Quantifies knockdown efficiency | SYBR Green (general) or Taqman/ Molecular Beacons (specific) [96] |
| Primer Sets | Amplifies gene of interest for quantification | Designed from Vg cDNA sequence [48] [2] |
| Reference Genes | Normalizes expression data | Alpha-tubulin (AT), housekeeping genes (HKGs) [14] |
Molecular Validation Workflow
The following diagram illustrates the core experimental workflow for verifying gene knockdown and assessing phenotypic effects, from initial gene silencing to final phenotypic analysis:
Comparative Phenotypic Outcomes of Vitellogenin Knockdown
Reproductive and Developmental Phenotypes
Vg knockdown consistently produces profound defects in oogenesis and embryogenesis across insect orders, though with species-specific variations in phenotypic severity:
Table 2: Reproductive and Developmental Phenotypes Following Vg Knockdown
| Species | Knockdown Efficiency | Oogenesis Defects | Embryonic Viability | Additional Reproductive Effects |
|---|---|---|---|---|
| Anthonomus grandis(Cotton Boll Weevil) | ~90% transcript reduction [48] | Not affected [48] | Nearly 100% loss [48] | Aberrant embryo development [48] |
| Rhodnius prolixus(Kissing Bug) | Significant reduction (Vg1 & Vg2) [4] | Yolk-depleted eggs [4] | Drastically reduced [4] | Reduced RHBP uptake [4] |
| Anopheles gambiae(Mosquito) | Confirmed via RNAi [95] | Not specified [95] | Not specified [95] | Reduced parasite survival (TEP1-dependent) [95] |
| Apis mellifera(Honey Bee) | Persistent protein suppression [2] | Not primary focus [2] | Not primary focus [2] | Accelerated foraging onset [2] |
Behavioral and Physiological Phenotypes
Beyond reproduction, Vg knockdown induces significant alterations in behavior, lifespan, and immune function, demonstrating the protein's pleiotropic nature:
Table 3: Behavioral and Physiological Phenotypes Following Vg Knockdown
| Species | Behavioral Changes | Lifespan Effects | Immune & Other Functions |
|---|---|---|---|
| Apis mellifera(Honey Bee) | Earlier foraging onset, nectar specialization [2] | Reduced longevity [2] | Not specified |
| Temnothorax longispinosus(Ant) | Reduced brood care, increased nestmate care [14] | Not specified | Altered responsiveness to social cues [14] |
| Rhodnius prolixus(Kissing Bug) | Not specified | Increased in both males and females [4] | Not specified |
| Anopheles gambiae(Mosquito) | Not specified | Not specified | Enhanced TEP1 binding to parasites [95] |
Regulatory Networks and Functional Interactions
Vitellogenin operates within complex physiological networks, interacting with nutrient-sensing pathways, hormonal systems, and immune factors. The following diagram illustrates key molecular relationships between Vg and associated pathways based on experimental evidence:
Key regulatory interactions supported by experimental evidence:
- Vg and Juvenile Hormone Form a Feedback Loop: In honey bees, Vg and JH mutually suppress each other, creating a regulatory network that influences both reproductive status and behavioral maturation [2].
- Nutrient Sensing Pathways Regulate Vg Expression: The Target of Rapamycin (TOR) pathway, activated by amino acids from blood meals, initiates massive Vg synthesis in the mosquito fat body [95].
- Immune System Interactions: In Anopheles gambiae, Vg and the lipid transporter Lipophorin (Lp) reduce the efficiency of the antiparasitic factor TEP1, demonstrating a trade-off between reproduction and immunity [95].
- NF-κB Pathway Inhibition: The NF-κB factor REL1 inhibits Vg expression after an infectious blood meal in mosquitoes, creating a molecular link between immune activation and reproductive investment [95].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for Vitellogenin Functional Studies
| Reagent/Category | Specific Examples | Function in Experimental Design |
|---|---|---|
| RNAi Reagents | dsRNA (target-specific), dsiRNA [14] | Induces gene-specific silencing [48] |
| RNA Extraction Kits | TRIzol Reagent, RNeasy Micro Kit [48] | Isolves high-quality total RNA [48] |
| Reverse Transcription Kits | Superscript III First-Strand Synthesis [48] | Generates cDNA from RNA templates [48] |
| qPCR Reagents | SYBR Green ROX Plus PCR Mix [48] | Enables real-time quantification of DNA [96] |
| Reference Genes | Alpha-tubulin (AT), Housekeeping genes (HKG) [14] | Normalizes qRT-PCR expression data [14] |
| Control dsRNA | GFP dsRNA [2] | Serves as handling disturbance control [2] |
Experimental data from multiple insect systems demonstrate that Vg knockdown produces a recognizable syndrome of phenotypic effects, though with species-specific variations. Core reproductive phenotypes include yolk-deficient oocytes and embryonic lethality, confirming Vg's fundamental role in egg provisioning. The pleiotropic behavioral and physiological effects—including altered behavioral timing, task specialization, lifespan modulation, and immune trade-offs—highlight the evolutionary co-option of this ancestral reproductive protein for social and life-history regulation. qRT-PCR validation remains essential for correlating the degree of gene silencing with phenotypic severity, enabling precise molecular explanations for observed phenotypic outcomes. These consistent findings across evolutionarily diverse species underscore Vg's central position in the network of genes coordinating reproduction, behavior, and physiology in insects.
Vitellogenin (Vg), a conserved yolk precursor protein, serves as a critical target for gene knockdown experiments to understand its diverse roles in reproduction, behavior, immunity, and aging across species. This comparative guide synthesizes experimental data from RNA interference (RNAi) studies targeting Vg and Vg-like genes in insects and crustaceans, providing a framework for researchers utilizing qRT-PCR validation in gene function studies. The functional diversification of Vg genes, arising from gene duplication events and subsequent subfunctionalization, has resulted in a complex landscape where Vg knockdown produces species-specific and genotype-dependent phenotypic outcomes [97]. This analysis objectively compares methodological approaches, phenotypic consequences, and technical considerations for Vg manipulation, with particular emphasis on appropriate normalization strategies for qRT-PCR in non-model organisms.
Functional Diversity of Vg and Vg-Like Genes
The Vg gene family belongs to the large lipid transfer protein (LLTP) superfamily and has undergone significant diversification across taxa. Conventional Vg is primarily associated with vitellogenesis and oocyte development, while Vg-like genes have acquired novel functions in immunity, oxidative stress response, and social behavior regulation [97]. In the brown planthopper (Nilaparvata lugens), researchers have identified a conventional Vg (NlVg) and two Vg-like genes (NlVg-like1 and NlVg-like2) with distinct functions. Phylogenetic analyses reveal that these Vg-like genes do not cluster with conventional insect Vgs associated with vitellogenesis, indicating functional divergence [97].
Gene duplication events have been a crucial driver of Vg functional diversification. Insects exhibit remarkable variation in Vg copy numbers, ranging from a single copy in honeybees (Apis mellifera) and silkworms (Bombyx mori) to five copies in mosquitoes (Aedes aegypti) and ants (Linepithema humile) [97]. These duplication events have enabled functional specialization, exemplified by the three Vg homologs in Hymenoptera (Vg-likeA, Vg-likeB, and Vg-likeC) that have acquired roles in determining longevity, oxidative stress response, and other non-vitellogenic functions [97].
Comparative Analysis of Vg Knockdown Phenotypes
Table 1: Comparative Effects of Vg and Vg-like Gene Knockdown Across Species
| Species | Gene Target | Knockdown Method | Key Phenotypic Outcomes | Molecular Validation Method |
|---|---|---|---|---|
| Nilaparvata lugens (Brown Planthopper) | NlVg | dsRNA (abdominal injection) | Essential for oocyte development and nymph development; lethal effects | qRT-PCR with fat body-specific expression analysis [97] |
| Nilaparvata lugens (Brown Planthopper) | NlVg-like1 | dsRNA (abdominal injection) | 18% offspring embryo failure: death before eggshell emergence; role in late embryogenesis | Temporal expression profiling across developmental stages [97] |
| Nilaparvata lugens (Brown Planthopper) | NlVg-like2 | dsRNA (abdominal injection) | 65% egg hatch failure; role in nutrition during oocyte/embryonic development | Spatial expression analysis (primarily female adults) [97] |
| Temnothorax longispinosus (Ant) | Vg-like A | RNAi (fat body knockdown) | Reduced brood care, increased nestmate care; shifted social cue responsiveness | Behavioral assays with chemical cue responsiveness tests [21] |
| Apis mellifera (Honey Bee) - High Strain | Vg | dsRNA (abdominal injection) | Precocious foraging, decreased lifespan; increased oxidative susceptibility | JH titer measurements, behavioral maturation scoring [98] |
| Apis mellifera (Honey Bee) - Low Strain | Vg | dsRNA (abdominal injection) | Increased lifespan; no behavioral maturation effect; genotype-dependent response | Targeted gene expression (Ilp1, Ilp2, mnSOD) in fat body [98] |
| Apis mellifera (Honey Bee) | Vg & ultraspiracle (usp) | Double gene knockdown (dsRNA mixture) | Altered gustatory perception; disrupted Vg-JH feedback loop | Proboscis extension response (PER) assay [99] |
Table 2: Quantitative Comparison of Knockdown Efficacy and Phenotypic Severity
| Species/Gene Target | Knockdown Efficiency | Reproductive Impact | Developmental Impact | Behavioral Impact | Lifespan Effect |
|---|---|---|---|---|---|
| N. lugens NlVg | High (essential gene) | Severe (oocyte development failure) | Severe (nymph development impaired) | Not assessed | Not assessed |
| N. lugens NlVg-like1 | Moderate | Moderate (18% embryo failure) | Moderate (late embryogenesis) | Not assessed | Not assessed |
| N. lugens NlVg-like2 | Moderate | Severe (65% egg hatch failure) | Moderate (embryonic development) | Not assessed | Not assessed |
| T. longispinosus Vg-likeA | High | Not assessed | Not assessed | Strong (task switching) | Not assessed |
| A. mellifera (High Strain) Vg | High | Not assessed | Not assessed | Strong (precocious foraging) | Decreased |
| A. mellifera (Low Strain) Vg | High | Not assessed | Not assessed | None | Increased |
Experimental Protocols for Vg Knockdown and Validation
RNAi-Mediated Gene Knockdown
The primary method for Vg knockdown involves dsRNA synthesis and injection:
dsRNA Synthesis: Design primers using software such as Primer3. Utilize the RiboMax T7 RNA production system for in vitro transcription. Perform dsRNA purification through denaturation (85°C for 5 minutes) and renaturation (room temperature for 1 hour), followed by DNase I treatment, chloroform extraction, and isopropyl alcohol precipitation [99].
dsRNA Injection: Immobilize subjects by chilling (4°C for 1-2 minutes). Mount specimens and inject 3μL of dsRNA (concentration ~9-10 μg/μL) using a microsyringe with a 30G needle. For abdominal injection, insert the needle to the side of the abdomen to avoid internal organs, slowly expel dsRNA, and leave the needle in place for 4-5 seconds before withdrawal [99].
Double Gene Knockdown Strategies: For simultaneous knockdown of multiple genes, two approaches are effective:
- Single Injection: Mix dsRNA targeting both genes and inject simultaneously
- Two-Day Injection: Inject dsRNA for the first gene on day one, followed by dsRNA for the second gene into the same subjects on day two [99]
qRT-PCR Validation and Reference Gene Selection
qRT-PCR validation requires appropriate reference gene selection, particularly in non-model organisms where housekeeping gene expression may vary under experimental conditions:
Candidate Reference Genes: Common reference genes include alpha-tubulin (Atb), β-actin (Act), 18S ribosomal RNA (18S), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), elongation factor 1-beta (EF-1b), ubiquitin conjugating enzyme (UBC), histone H2A (H2A), TATA-box binding protein (TBP), and succinate dehydrogenase (SDH) [18].
Stability Validation: Evaluate reference gene stability using multiple algorithms (geNorm, NormFinder, BestKeeper) under specific experimental conditions. For chemical exposure studies in crustaceans, H2A and Act demonstrate high stability, while Atb shows significant variation across developmental stages [18].
Normalization Strategy: Use at least two validated reference genes for normalization to avoid incorrect results. For age-related studies in crustaceans, reference gene stability should be specifically validated across developmental stages due to notable changes during molting cycles [18].
Signaling Pathways and Molecular Networks
The Vg gene network interacts with several conserved signaling pathways, with significant cross-talk between Vg, juvenile hormone (JH), and insulin signaling. The following diagram illustrates the core regulatory feedback loop between Vg and JH, and their combined impact on behavior and physiology:
Diagram 1: Vg-JH Regulatory Network
The experimental workflow for Vg knockdown studies involves a multi-stage process from gene targeting to phenotypic analysis, as illustrated below:
Diagram 2: Experimental Workflow for Vg Knockdown Studies
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for Vg Knockdown Studies
| Reagent/Resource | Function/Application | Specifications | Example Use Cases |
|---|---|---|---|
| T7 RiboMax Express System | In vitro transcription for dsRNA synthesis | Produces high-yield, pure dsRNA | dsRNA synthesis for Vg and Vg-like gene knockdown [99] |
| qRT-PCR Reference Genes | Normalization of gene expression data | Must be validated for specific species and conditions | H2A and Act for chemical exposure in crustaceans; multiple genes for developmental stages [18] |
| Gene-Specific Primers | Amplification of target sequences | Designed with software such as Primer3; ~80-100 bp product size | Vg, Vg-like genes, and reference gene amplification [99] [18] |
| Microsyringe (Hamilton) | Precise dsRNA delivery | 30G needle for abdominal injection | RNAi in insects and small crustaceans [99] |
| Color Contrast Analyzer | Accessibility verification for diagrams | WCAG 2.1 AA compliance (4.5:1 ratio for text) | Ensuring readability of research visuals [100] [101] |
Discussion and Research Implications
The comparative analysis of Vg knockdown across species reveals several critical patterns. First, the phenotypic consequences of Vg manipulation are highly context-dependent, influenced by genetic background, ecological niche, and social structure. In the brown planthopper, conventional Vg is essential for reproduction, while Vg-like genes have subfunctionalized for embryogenesis and nutritional functions [97]. In social insects, Vg and Vg-like genes have been co-opted for behavioral regulation, exemplified by Vg-like A's role in task specialization in ants [21].
Second, genetic background significantly influences responses to Vg manipulation, as demonstrated by the divergent effects of Vg knockdown in honey bee strains. In the high strain (characterized by low Vg titers), lifespan increases following Vg knockdown, suggesting artificial selection has driven the expansion of alternative maintenance mechanisms [98]. This genotype-dependent response underscores the importance of considering genetic variation when interpreting knockdown experiments.
Methodologically, rigorous qRT-PCR validation using appropriate reference genes is essential for accurate interpretation of Vg knockdown studies. Research on brackish water flea (Diaphanosoma celebensis) demonstrates that reference gene stability varies significantly with chemical exposure type and developmental stage [18]. This highlights the necessity of validating reference genes for specific experimental conditions rather than relying on presumed "housekeeping" genes.
The functional diversification of Vg and Vg-like genes across species presents both challenges and opportunities for comparative studies. While the core Vg structure and its role in reproduction appear conserved, gene duplication events have enabled lineage-specific functional specialization. This evolutionary flexibility makes Vg an intriguing model for studying gene family evolution and functional diversification, particularly in the context of social insect evolution where Vg and Vg-like genes have been co-opted for behavioral regulation [97] [21].
In molecular biology research, qRT-PCR has become a standard tool for validating gene knockdown efficacy. However, an overreliance on mRNA quantification presents a significant limitation: transcriptional silencing does not always translate to functional protein reduction or phenotypic manifestation. This guide compares standalone qRT-PCR validation against integrated multi-method approaches, using vitellogenin (Vg) knockdown research as a case study to demonstrate why combining proteomic and histological data provides a more comprehensive validation framework.
The Validation Spectrum: Methodologies Compared
| Methodology | Target Level | Key Readout | Temporal Context | Limitations |
|---|---|---|---|---|
| qRT-PCR | mRNA | Transcript abundance | Early post-knockdown | Does not confirm protein-level reduction |
| Western Blot | Protein | Protein presence/quantity | Mid-stage validation | Cannot assess spatial distribution or function |
| Proteomics | Protein network | Global protein expression | Systems-level analysis | Complex data analysis; resource intensive |
| Immunohistochemistry | Protein in tissue | Protein localization & context | Spatial preservation | Semi-quantitative; tissue quality dependent |
| Phenotypic Analysis | Organism/Tissue | Functional consequence | Ultimate validation | May not directly link to molecular target |
Vitellogenin Knockdown: A Case Study in Multi-Method Validation
Vitellogenin, a conserved yolk protein precursor, serves as an excellent model for validation methodology comparison due to its well-characterized role in reproduction across insect species. The tables below summarize key experimental findings from Vg knockdown studies across different model organisms.
Table 1: Vitellogenin Knockdown Efficiency Across Validation Methods
| Study Organism | qRT-PCR Reduction | Protein Reduction | Histological Phenotype | Functional Outcome | Citation |
|---|---|---|---|---|---|
| Red palm weevil | 95-99% (15-25 dpi) | Dramatic failure of Vg protein expression | Atrophied ovaries, no oogenesis | Complete egg hatch failure | [7] |
| Cotton boll weevil | ~90% transcript reduction | Not quantified | Aberrant embryo phenotypes | ~100% egg viability loss | [102] |
| Ant (T. longispinosus) | Vg-like A knockdown | Not directly quantified | Not applicable | Reduced brood care, increased nestmate care | [21] |
| Honey bee | Well-established protocol | Confirmed via proteomics | Not applicable | Accelerated behavioral maturation | [3] |
Table 2: Temporal and Spatial Resolution of Validation Methods in Vg Research
| Validation Method | Detection Timeframe | Tissue Specificity | Quantitative Potential | Phenotypic Correlation |
|---|---|---|---|---|
| qRT-PCR | Days post-knockdown | Tissue extracts possible | High (absolute) | Indirect |
| Western Blot | Days to weeks | Tissue extracts possible | Semi-quantitative | Moderate |
| Proteomic Analysis | Single timepoint snapshot | Tissue/cell type specific | High (relative) | Pathway-level |
| Histology/IHC | Weeks (morphological) | Preserved spatial context | Low to moderate | Direct visual correlation |
Experimental Protocols for Holistic Validation
Protocol 1: qRT-PCR Validation of Vitellogenin Knockdown
Principle: Quantify reduction in Vg mRNA levels following RNAi treatment using sequence-specific primers.
Procedure:
- RNA Extraction: Isolate total RNA from fat body tissue (primary Vg production site) using TRIzol protocol
- cDNA Synthesis: Convert 1-2μg RNA using reverse transcriptase with oligo(dT) or random hexamers
- qPCR Amplification:
- Reaction mix: 0.5μL 20X SYBR green Mastermix, 1μL each primer (25pmol/μL), 2μL diluted cDNA, 20.5μL DEPC water
- Cycling: 94°C for 4min, 35 cycles of (94°C for 20s, 60°C for 30s, 72°C for 30s)
- Data Analysis: Calculate relative expression using 2^(-ΔΔCt) method with 18S rRNA as internal control [103]
Protocol 2: Proteomic Validation via iTRAQ-Based Quantification
Principle: Compare global protein expression between treated and control groups using isobaric tags.
Procedure:
- Protein Extraction:
- Homogenize tissue in PASP lysis buffer (100mmol/L ammonium bicarbonate, 8mol/L urea, pH8)
- Centrifuge at 12,000×g, 4°C for 15min, collect supernatant
- Reduce with 10mmol/L DTT (56°C, 1h), alkylate with IAM (room temp, dark, 1h)
- Acetone precipitate at -20°C for 2h, collect pellet [104]
Digestion and Labeling:
- Digest with trypsin (37°C, 4h) at 1:50 enzyme:protein ratio
- Label with iTRAQ reagents (5.5μL per sample, shake 2h)
- Terminate reaction with 100μL 50mmol/L Tris-HCl (pH8) [104]
LC-MS/MS Analysis:
- Fractionate using reverse-phase chromatography
- Analyze by tandem mass spectrometry
- Identify and quantify proteins using database searching
Protocol 3: Histological Validation of Ovarian Phenotypes
Principle: Visualize morphological consequences of Vg knockdown in reproductive tissues.
Procedure:
- Tissue Collection and Fixation: Dissect ovaries in physiological saline, fix in 4% paraformaldehyde (4°C, 24h)
- Processing and Sectioning:
- Dehydrate through graded ethanol series
- Clear with xylene, embed in paraffin
- Section at 5μm thickness
- Staining and Analysis:
- Deparaffinize and rehydrate sections
- Stain with Hematoxylin and Eosin (H&E)
- For specific detection: Perform immunohistochemistry with Vg antibodies [15]
- Image and analyze oocyte development, vitellogenin uptake impairment
Visualizing Experimental Workflows
Integrated Validation Pathway
Vitellogenin Disruption Consequences
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Vitellogenin Knockdown Validation Studies
| Reagent/Category | Specific Examples | Research Function | Application Notes |
|---|---|---|---|
| RNAi Reagents | dsRNA targeting Vg gene sequence | Gene silencing induction | Species-specific design critical for efficacy |
| qRT-PCR Components | SYBR Green Mastermix, Vg-specific primers, 18S rRNA controls | mRNA quantification | Optimize primer efficiency (90-110%) |
| Proteomic Tools | iTRAQ labels, trypsin, C18 columns, mass spectrometry systems | Protein identification and quantification | Requires specialized instrumentation |
| Histological Reagents | Vg antibodies, H&E stains, paraformaldehyde, paraffin | Tissue preservation and visualization | Antibody specificity validation required |
| Positive Controls | Known Vg-expressing tissues, validated knockdown models | Experimental validation | Enables protocol standardization |
The integration of proteomic and histological data with traditional qRT-PCR validation represents a paradigm shift in gene knockdown research. While qRT-PCR provides essential initial confirmation of transcriptional silencing, the case studies in vitellogenin research demonstrate that functional protein reduction and phenotypic outcomes require additional validation layers. Proteomic approaches confirm the actual reduction of target proteins and identify potential compensatory mechanisms, while histological analysis provides spatial context and morphological correlation. This multi-dimensional validation framework not only strengthens experimental conclusions but also reveals broader biological insights, ultimately advancing the reliability and translational potential of gene knockdown research.
Adhering to MIQE Guidelines and CR Assay Standards for Reproducible Research
The Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines establish a standardized framework designed to ensure the reproducibility, reliability, and transparency of qPCR experiments [105] [106]. First published in 2009 and recently updated to MIQE 2.0, these guidelines provide researchers with clear recommendations for every stage of the qPCR workflow, from experimental design and sample handling to data analysis and reporting [107] [108]. The primary goal is to address the widespread methodological failures and reporting deficiencies that have undermined the credibility of qPCR data in scientific literature [108].
In the specific context of vitellogenin knockdown research, where qPCR is frequently employed to measure gene expression changes following genetic manipulation, adherence to MIQE principles becomes particularly crucial. Studies investigating genes such as vitellogenin-2 (vit-2) in C. elegans have demonstrated significant implications for lipid metabolism and lifespan regulation [12]. Without rigorous experimental design and validation, conclusions drawn from such research may be unreliable. The MIQE 2.0 update specifically addresses emerging complexities in qPCR applications, offering tailored guidance for contemporary research needs while maintaining the core principle that transparent reporting of all experimental details is essential for scientific integrity [107].
Core Principles of MIQE 2.0 Guidelines
Fundamental Requirements for Transparent Reporting
The revised MIQE 2.0 guidelines emphasize several foundational principles that researchers must address to ensure qPCR data credibility. These requirements have been updated to reflect technological advancements while maintaining the rigorous standards established in the original publication [107] [106]. A core emphasis is placed on the comprehensive documentation of all experimental details, enabling both reviewers and other researchers to critically evaluate and reproduce the results [107].
Key advancements in MIQE 2.0 include clarified recommendations for sample handling, assay design, and validation, along with enhanced guidance for qPCR data analysis [107]. The guidelines specifically stress that instrument manufacturers should enable raw data export to facilitate thorough analysis and re-evaluation by manuscript reviewers [107]. Furthermore, the updated guidelines streamline reporting requirements to encourage researcher compliance without imposing undue burden, thereby promoting more widespread adoption of these critical standards [107].
Essential MIQE Checklist Components
Table 1: Essential MIQE Checklist Components for Vitellogenin Knockdown Research
| Category | Requirement | Application to Vitellogenin Research |
|---|---|---|
| Sample Quality | RNA quality/integrity assessment | Document RIN values for all C. elegans samples |
| Assay Validation | PCR efficiency, linear dynamic range | Validate primers for all vitellogenin genes (vit-1 to vit-6) |
| Specificity | Amplicon sequence disclosure | Provide context sequences for all target and reference genes |
| Data Analysis | Cq conversion to efficiency-corrected quantities | Report normalized vitellogenin expression with prediction intervals |
| Normalization | Use of validated reference genes | Employ multiple stable reference genes (e.g., pmp-3 in C. elegans) |
| Limits of Detection | LOQ and LOD determination | Establish detection limits for each vitellogenin assay |
Implementing MIQE Standards in Vitellogenin Research
Experimental Design and Sample Preparation
Implementing MIQE guidelines in vitellogenin knockdown research begins with rigorous experimental design and sample preparation. In the cited C. elegans study investigating PRY-1/Axin regulation of vitellogenin-2, researchers maintained standardized growth conditions, culturing worms at 20°C on nematode growth media plates seeded with E. coli OP50 [12]. This consistency in environmental conditions is crucial for minimizing biological variability that could compromise qPCR results.
For sample collection, the researchers employed bleach synchronization to obtain developmentally staged worms, followed by RNA extraction using Tri-reagent according to the manufacturer's protocol [12]. The MIQE guidelines emphasize that RNA quality must be properly assessed before proceeding with cDNA synthesis, as the integrity of starting material fundamentally impacts result reliability [108]. The cDNA was then synthesized using oligo(dT) primers and the SensiFAST cDNA kit, ensuring the reverse transcription step was performed under standardized conditions [12]. This attention to methodological detail in the pre-amplification phase represents exactly the type of rigorous approach that MIQE guidelines promote.
Assay Design and Validation for Vitellogenin Targets
Proper assay design and validation are cornerstone requirements of the MIQE guidelines. In vitellogenin research, this involves developing and validating target-specific assays for genes of interest. The C. elegans study provided a comprehensive list of primers for vitellogenin genes (vit-1 through vit-6), along with the reference gene pmp-3 [12]. According to MIQE standards, each of these assays requires validation to establish key parameters.
Table 2: Required Assay Validation Parameters per MIQE Guidelines
| Parameter | Requirement | Validation Method |
|---|---|---|
| Amplification Efficiency | 90-110% | Standard curve from serial dilutions |
| Linear Dynamic Range | At least 5 logs | Plot Cq vs. log template concentration |
| Limit of Quantification | Lowest quantity with stated accuracy | Point where Cq stops being co-linear with input |
| Specificity | Single amplification product | Melt curve analysis |
| Precision | Repeatability and reproducibility | Intra- and inter-assay variance |
The limit of quantification is particularly critical, defined as the lowest amount of measurand that can be quantitatively determined with stated acceptable precision and accuracy [109]. For qRT-PCR assays, the LOQ can be considered the point at which the Cq value stops being co-linear with the template concentration, representing the bottom of the linear dynamic range [109].
Reference Gene Validation in Vitellogenin Studies
Appropriate normalization using validated reference genes is essential for accurate gene expression quantification in vitellogenin research. The MIQE guidelines emphasize that reference genes must be stable under specific experimental conditions, as commonly used housekeeping genes can vary significantly [18] [108]. The C. elegans study utilized pmp-3 as a reference gene for normalizing vitellogenin expression data [12].
Research on brackish water fleas demonstrated that reference gene stability can vary dramatically under different experimental conditions, with distinct patterns observed during chemical exposure versus aging [18]. This underscores the MIQE principle that reference genes must be validated for each specific experimental context rather than assumed to be stable. Comprehensive validation typically involves assessing multiple candidate reference genes using algorithms such as geNorm, NormFinder, and BestKeeper, then selecting the most stable genes for normalization [18]. The MIQE guidelines recommend using at least two validated reference genes to ensure accurate normalization [107].
Signaling Pathways and Experimental Workflows
PRY-1/Axin to Vitellogenin-2 Signaling Pathway
The relationship between PRY-1/Axin and vitellogenin-2 represents a key signaling pathway in C. elegans that regulates lipid metabolism and lifespan. The following diagram illustrates this pathway and its functional outcomes:
This pathway demonstrates how PRY-1/Axin negatively regulates vitellogenin-2 expression, which in turn modulates lipid levels and lifespan in C. elegans [12]. Knockdown of vit-2 during adulthood significantly rescued lipid levels in pry-1 mutants (almost 2-fold) and markedly rescued the lifespan defect (102% increase in mean lifespan) [12]. These findings establish vit-2 as functioning downstream of pry-1 to regulate both lipid metabolism and aging.
qPCR Experimental Workflow for Vitellogenin Analysis
The complete experimental workflow for vitellogenin expression analysis involves multiple critical steps from sample preparation to data reporting, each requiring careful attention to MIQE guidelines:
This workflow illustrates the sequential process for vitellogenin expression analysis, highlighting critical stages where MIQE guidelines provide specific recommendations to ensure data reliability and reproducibility.
Research Reagent Solutions for MIQE-Compliant Vitellogenin Studies
Essential Reagents and Kits
Table 3: Research Reagent Solutions for MIQE-Compliant Vitellogenin Studies
| Reagent/Kits | Specific Product Example | Function in Vitellogenin Research | MIQE Compliance Aspect |
|---|---|---|---|
| RNA Extraction | Tri-reagent (Sigma-Aldrich T9424) | Isolation of total RNA from C. elegans | Documentation of RNA quality and integrity |
| cDNA Synthesis | SensiFAST cDNA Kit (BIO-65054) | Reverse transcription with oligo(dT) primers | Standardized reverse transcription conditions |
| qPCR Master Mix | SensiFAST SYBR Green Kit (BIO-98005) | Fluorescent detection of amplification | Documentation of reaction chemistry |
| Reference Genes | pmp-3 primers | Normalization of gene expression data | Use of validated reference genes |
| Target Assays | vit-1 to vit-6 primers | Quantification of vitellogenin expression | Target-specific validation data |
Instrumentation and Analysis Tools
Implementation of MIQE guidelines requires appropriate instrumentation and software tools. The C. elegans vitellogenin study utilized a CFX 96 BioRad cycler for qPCR amplification [12], which enables the precise thermal cycling and fluorescence detection necessary for reliable Cq determination. For data analysis, the researchers employed Bio-Rad CFX manager software for Ct value calculation and statistical analysis [12].
The MIQE guidelines specifically recommend that instrument manufacturers enable raw data export to facilitate independent re-analysis [107], a feature that should be considered when selecting instrumentation for vitellogenin studies. For specialized analysis, including reference gene validation, tools such as GeNorm, NormFinder, and BestKeeper provide algorithms for assessing gene stability [18], while comprehensive web-based tools like RefFinder integrate multiple approaches to rank candidate reference genes [18].
Comparative Analysis of qPCR Validation Methods
Validation Parameter Implementation
Successful implementation of MIQE guidelines requires thorough understanding and application of qPCR validation methods. The following table compares key validation parameters and their application in vitellogenin research:
Table 4: qPCR Validation Methods and Applications in Vitellogenin Research
| Validation Parameter | Calculation Method | Acceptance Criteria | Application in Vitellogenin Study |
|---|---|---|---|
| Amplification Efficiency | Standard curve from serial dilutions | 90-110% | Efficiency values ranged 90.8-106.7% [18] |
| Linear Dynamic Range | Plot of Cq vs. log template concentration | Minimum 5 orders of magnitude | Established for each vitellogenin assay |
| Limit of Quantification | Point where linearity is lost | Lowest point in dynamic range | Determined for each target [109] |
| Precision (Repeatability) | Intra-assay variance | CV < 5% | All qPCR reactions performed in triplicate [12] |
| Specificity | Melt curve analysis | Single peak | Unimodal melting curves confirmed [18] |
Comparison of Reference Gene Validation Tools
Each reference gene validation method employs distinct algorithms and provides complementary information for assessing gene stability:
Table 5: Comparison of Reference Gene Validation Algorithms
| Validation Tool | Algorithm Basis | Output | Advantages | Limitations |
|---|---|---|---|---|
| geNorm | Pairwise comparison of expression ratios | M-value (stability measure) | Determines optimal number of reference genes | May select co-regulated genes |
| NormFinder | Intra- and inter-group variation analysis | Stability value | Resistant to co-regulation | Does not suggest number of genes needed |
| BestKeeper | Pairwise correlation analysis | Standard deviation | Works well with small gene sets | Requires high expression stability |
| RefFinder | Comprehensive integration of multiple tools | Comprehensive ranking | Combines advantages of all methods | Web-based, requires internet access |
Adherence to MIQE guidelines represents a critical foundation for generating reliable, reproducible data in vitellogenin knockdown research and qPCR studies broadly. The recently published MIQE 2.0 guidelines provide updated recommendations that address emerging technologies and evolving applications while maintaining core principles of transparency and methodological rigor [107]. As evidenced by the C. elegans vitellogenin study, comprehensive reporting of experimental details—including sample handling, assay validation, reference gene selection, and data analysis methods—enables proper evaluation and repetition of experimental findings [12].
The scientific community faces ongoing challenges in implementing these standards, with continued deficiencies observed in experimental transparency, assay validation, and data reporting despite widespread awareness of MIQE [108]. However, the consequences of methodological failures are particularly significant in vitellogenin research, where conclusions may impact understanding of lipid metabolism, aging, and disease mechanisms. By embracing the MIQE 2.0 framework as a practical guide rather than a burdensome requirement, researchers can significantly enhance the credibility of their findings and contribute to a more robust, reproducible scientific literature.
Conclusion
The successful qRT-PCR validation of vitellogenin knockdown hinges on a integrated approach that combines a deep understanding of Vg biology, a rigorously optimized and troubleshooted molecular workflow, and a comprehensive validation strategy that links molecular data to clear phenotypic outcomes. Adherence to established guidelines like MIQE is non-negotiable for generating reliable, reproducible data. The implications of this research are vast, enabling the development of targeted RNAi-based biocontrols for agricultural pests and providing powerful tools for deciphering complex genetic networks governing reproduction, aging, and social behavior. Future directions should focus on improving in vivo dsRNA delivery methods, exploring the regulatory functions of Vg fragments, and translating these research assays into clinically validated diagnostic tools.
References
- 1. Vitellogenin - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/vitellogenin]
- 2. The Gene vitellogenin Has Multiple Coordinating Effects on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1808115/]
- 3. Evidence for vitellogenin DNA‐binding in honey bees - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12432429/]
- 4. Functional characterization of vitellogenin unveils novel ... [https://www.sciencedirect.com/science/article/abs/pii/S0965174825000451]
- 5. Vitellogenin plays a role in regulating honey bee swarming [https://www.nature.com/articles/s41598-025-20547-z]
- 6. Downregulation of vitellogenin gene activity increases the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2398700/]
- 7. Silencing of vitellogenin gene contributes to the promise of ... [https://www.nature.com/articles/s41598-021-01159-9]
- 8. CRISPR/Cas9-Mediated Vitellogenin Receptor Knockout ... [https://www.frontiersin.org/journals/physiology/articles/10.3389/fphys.2019.01585/full]
- 9. Vitellogenin Receptor as a Target for Tick Control [https://pmc.ncbi.nlm.nih.gov/articles/PMC6537121/]
- 10. Genome-Wide Identification of Vitellogenin Gene Family and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10815947/]
- 11. Molecular characterization and CRISPR/Cas9 validation of ... [https://www.sciencedirect.com/science/article/abs/pii/S0378111924008060]
- 12. Vitellogenin-2 acts downstream of PRY-1/Axin to regulate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7382951/]
- 13. Genomic disturbance of vitellogenin 2 (vtg2) leads to ... [https://www.nature.com/articles/s41598-023-46148-2]
- 14. Vitellogenin-like A–associated shifts in social cue ... [https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.2005747]
- 15. Vitellogenin receptor mediates heat adaptability of oocyte ... [https://www.nature.com/articles/s41467-025-59035-3]
- 16. Vitellogenin-like A-associated Shifts in Social Cue ... [https://pubmed.ncbi.nlm.nih.gov/29874231/]
- 17. Expression and Role of Vitellogenin Genes in Ovarian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9143374/]
- 18. Validation of reference genes for quantitative real-time ... [https://www.nature.com/articles/s41598-021-03098-x]
- 19. Vitellogenin - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/neuroscience/vitellogenin]
- 20. Vitellogenin, juvenile hormone, insulin signaling, and queen ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1852330/]
- 21. Vitellogenin-like A–associated shifts in social cue ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5991380/]
- 22. Disruption of vitellogenin gene function in adult honeybees by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC149360/]
- 23. Single-cell transcriptomic analysis of honeybee brains ... [https://www.sciencedirect.com/science/article/pii/S2589004222009154]
- 24. Consensus guidelines for the validation of qRT-PCR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8792405/]
- 25. Quantitative PCR validation for research scientists: Part 1 of 2 [https://virologyresearchservices.com/2023/01/13/quantitative-pcr-validation-part-1-of-2/]
- 26. Development and validation of a qPCR assay for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12536431/]
- 27. Evaluation and validation of reference genes for RT-qPCR ... [https://www.nature.com/articles/s41598-025-22650-7]
- 28. Quantitative PCR primer design affects quantification of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6662389/]
- 29. RNase If -treated quantitative PCR for dsRNA quantitation of ... [https://bmcbiotechnol.biomedcentral.com/articles/10.1186/s12896-018-0413-6]
- 30. SIGS vs HIGS: a study on the efficacy of two dsRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6859480/]
- 31. SIGS vs HIGS: a study on the efficacy of two dsRNA ... [https://pubmed.ncbi.nlm.nih.gov/31603277/]
- 32. A comparative analysis of five methods for gene silencing [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0180654]
- 33. RNA interference-mediated knockdown of genes involved ... [https://www.frontiersin.org/journals/insect-science/articles/10.3389/finsc.2023.1283334/full]
- 34. Purification and characterisation of dsRNA using ion pair ... [https://www.sciencedirect.com/science/article/pii/S0021967316317150]
- 35. Intraperitoneal injection [https://en.wikipedia.org/wiki/Intraperitoneal_injection]
- 36. Microinjection - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/microinjection]
- 37. Microinjection: Definition, Principle, Steps, Uses [https://microbenotes.com/microinjection/]
- 38. Effects of Formula Product Injection on Hatching Parameters ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11720732/]
- 39. Impact of vitellogenin based dsRNA feeding on reproductive ... [https://jksus.org/impact-of-vitellogenin-based-dsrna-feeding-on-reproductive-biology-of-red-palm-weevil-rhynchophorus-ferrugineus-olivier-coleoptera-dryophthoridae-under-laboratory-conditions/]
- 40. In ovo applications in poultry: A review [https://www.sciencedirect.com/science/article/pii/S0032579119303451]
- 41. qPCR and qRT-PCR analysis: Regulatory points to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7786041/]
- 42. Choosing the Right Color PCR Plate [https://gmpplastic.com/blogs/useful-articles-on-lab-supplies-faq-section/choosing-the-right-color-pcr-plate?srsltid=AfmBOorVns29RAj5pViTOnwucQdzPFj1IyPABrBA5FCChsc5pEsxRv_e]
- 43. PCR Plates and qPCR Plates [https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/thermo-scientific-pcr/pcr-plates-and-qpcr-plates.html]
- 44. Advancing quantitative PCR with color cycle multiplex ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11417387/]
- 45. Selection and Validation of Stable Reference Genes for RT ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12112379/]
- 46. Selection and validation of reference genes for RT-qPCR ... [https://www.nature.com/articles/s41598-025-02951-7]
- 47. Selection and Validation of Reference Genes for qRT-PCR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9408421/]
- 48. Vitellogenin knockdown strongly affects cotton boll weevil egg ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4936639/]
- 49. Impact of vitellogenin based dsRNA feeding on ... [https://www.sciencedirect.com/science/article/pii/S1018364723001635]
- 50. Selection and Validation of Reference Genes for qRT-PCR ... [https://www.mdpi.com/2311-7524/9/11/1176]
- 51. Selection and evaluation of reference genes for qRT-PCR ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1500043/full]
- 52. Midgut-specific vitellogenin-1 is involved in the negative ... [https://www.sciencedirect.com/science/article/pii/S1877959X25000445]
- 53. Quantitative Real-Time Analysis of Differentially Expressed ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8872078/]
- 54. Development and analytical validation of a multiplex ... [https://www.sciencedirect.com/science/article/pii/S1936523325002591]
- 55. Comparative transcriptome analysis provides a glance into the ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11459-3]
- 56. Comparative Analyses of Reproductive Caste Types ... [https://www.mdpi.com/1422-0067/24/24/17130]
- 57. Mining, validating, and quantifying circular RNA ... [https://www.nature.com/articles/s41598-025-05652-3]
- 58. Top Ten Most Common Real-Time qRT - PCR Pitfalls [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/rtpcr-analysis/general-articles/ten-most-common-real-time-qrt-pcr-pitfalls.html]
- 59. guide - 5 tips for best Primer results design PCR [https://the-dna-universe.com/2022/09/05/primer-design-guide-the-top-5-factors-to-consider-for-optimum-performance/]
- 60. Rules and Tips for PCR & qPCR Primer | IDT Design [https://www.idtdna.com/page/support-and-education/decoded-plus/how-to-design-primers-and-probes-for-pcr-and-qpcr/]
- 61. PrimerQuest PCR & qPCR Primer Design Tool | IDT [https://www.idtdna.com/pages/tools/primerquest]
- 62. Primer designing tool - NCBI - NIH [https://www.ncbi.nlm.nih.gov/tools/primer-blast/]
- 63. qPCR Primer & Probe Design Tool [https://eurofinsgenomics.eu/en/ecom/tools/qpcr-assay-design/]
- 64. Real-time PCR (TaqMan) Primer and Probes Design Tool [https://www.genscript.com/tools/real-time-pcr-taqman-primer-design-tool]
- 65. qPCR optimization in focus: tips and tricks for excellent ... [https://www.genaxxon.com/shop/en/blog/qpcr-optimization-in-focus-tips-and-tricks-for-excellent-analyses]
- 66. What should the final primer concentration be in my PCR? [https://www.neb.com/en-us/faqs/2023/08/02/what-should-the-final-primer-concentration-be-in-my-pcr?srsltid=AfmBOoraoxFIgUzY1FuK3f7jsSyNaixGy-GneBPJTQ0LnfvqJ4VHKB9z]
- 67. Optimization Tips for Luna® One-Step RT-qPCR [https://www.neb.com/en-us/tools-and-resources/usage-guidelines/optimization-tips-for-luna-one-step-rt-qpcr?srsltid=AfmBOopEadmk3vO0vKBjyWR6tkP4-GxGjhjnT-cRJgPkwAWnGrUP3Jo6]
- 68. An optimized protocol for stepwise optimization of real-time ... [https://www.nature.com/articles/s41438-021-00616-w]
- 69. Top Ten Pitfalls in Quantitative Real-time PCR Primer ... [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/rtpcr-analysis/general-articles/top-ten-pitfalls-in-quantitative-real-time-pcr-primer.html]
- 70. Comparison of Seven Commercial TaqMan Master Mixes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7810389/]
- 71. Silencing of vitellogenin gene contributes to the promise of controlling... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8568968/]
- 72. Biomarkers for Monitoring Pre-Analytical Quality Variation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4219744/]
- 73. The importance of sample quality for qPCR [https://www.europeanpharmaceuticalreview.com/article/983/article-1-the-importance-of-sample-quality-for-qpcr/]
- 74. Methods of RNA Quality Assessment [https://www.promega.com/resources/pubhub/methods-of-rna-quality-assessment/]
- 75. RNA quantification and quality control methods [https://www.qiagen.com/us/knowledge-and-support/knowledge-hub/bench-guide/rna/rna-analysis/quantification-and-analysis-of-rna]
- 76. Is Your RNA Intact? Methods to Check RNA Integrity [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/rna-isolation/tech-notes/is-your-rna-intact.html]
- 77. Five Steps to Optimal cDNA Synthesis [https://www.thermofisher.com/br/en/home/life-science/pcr/reverse-transcription/5steps-cDNA.html]
- 78. Use and Misuse of Cq in qPCR Data Analysis and Reporting [https://pmc.ncbi.nlm.nih.gov/articles/PMC8229287/]
- 79. Effects of Double-Stranded RNA Degrading Nucleases on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11857036/]
- 80. Differential RNAi efficacy of siRNA and dsRNA targeting ... [https://www.frontiersin.org/journals/insect-science/articles/10.3389/finsc.2025.1574585/full]
- 81. Engineered RNA nanostructures for scalable and efficient ... [https://www.sciencedirect.com/science/article/abs/pii/S0167779925003129]
- 82. Engineered RNA nanostructures for scalable and efficient ... [https://pubmed.ncbi.nlm.nih.gov/40858461/]
- 83. Non-cationic carriers for siRNA delivery: Current progress ... [https://www.sciencedirect.com/science/article/abs/pii/S0168365925009769]
- 84. Evaluation of non-invasive dsRNA delivery methods for the ... [https://ui.adsabs.harvard.edu/abs/2025JPesS..98..581G/abstract]
- 85. Amplification of the No Template Control (NTC) [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/real-time-pcr-basics/real-time-pcr-troubleshooting-tool/gene-expression-quantitation-troubleshooting/amplification-no-template-control.html]
- 86. Adjusting RT-qPCR conditions to avoid unspecific ... [https://www.sciencedirect.com/science/article/pii/S1201971220322839]
- 87. Selection and validation of RT-qPCR reference genes for multi ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12465390/]
- 88. Evaluation and validation of suitable reference genes for ... [https://www.nature.com/articles/s41598-024-61806-9]
- 89. Functional Elucidation of Vitellogenin receptor Activity in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12295193/]
- 90. qPCR and RT-qPCR [https://www.promega.com/resources/guides/nucleic-acid-analysis/qpcr-and-rt-qpcr-amplification/]
- 91. Quantitative or digital PCR? A comparative analysis for ... [https://www.sciencedirect.com/science/article/abs/pii/S0165993624001584]
- 92. A high-sensitivity qPCR method for detecting residual Vero ... [https://www.sciencedirect.com/science/article/abs/pii/S0166093425001107]
- 93. Validation and advantages of using novel RT-qPCR ... [https://www.nature.com/articles/s41598-022-17339-0]
- 94. Comparison of qRT-PCR and ddPCR for multi-strain ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1579797/full]
- 95. The Major Yolk Protein Vitellogenin Interferes with the Anti ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2907290/]
- 96. qRT-PCR to verify RNAi knockdown – Bytes of the Apple [https://blogs.reed.edu/bytes-of-the-apple/2018/10/23/84/]
- 97. Vitellogenin and Vitellogenin-Like Genes in the Brown ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6759490/]
- 98. Genotype effect on lifespan following vitellogenin knockdown [https://www.sciencedirect.com/science/article/abs/pii/S0531556514003544]
- 99. RNAi-mediated Double Gene Knockdown and Gustatory ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3846282/]
- 100. Elements must meet minimum color contrast ratio thresholds [https://dequeuniversity.com/rules/axe/4.8/color-contrast]
- 101. Color contrast - Accessibility - MDN Web Docs - Mozilla [https://developer.mozilla.org/en-US/docs/Web/Accessibility/Guides/Understanding_WCAG/Perceivable/Color_contrast]
- 102. Vitellogenin knockdown strongly affects cotton boll weevil ... [https://www.sciencedirect.com/science/article/abs/pii/S2214540016300226]
- 103. 2.6. Quantitative Real-Time PCR (RT-qPCR) Validation of ... [https://bio-protocol.org/exchange/minidetail?id=620793&type=30]
- 104. Comparative Proteomic Insights into the Immune ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12295233/]
- 105. MIQE Guidelines | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-assays/why-choose-taqman-assays/publish-real-time-pcr-results.html]
- 106. The MIQE guidelines: minimum information for publication ... [https://pubmed.ncbi.nlm.nih.gov/19246619/]
- 107. MIQE 2.0: Revision of the Minimum Information for ... [https://pubmed.ncbi.nlm.nih.gov/40272429/]
- 108. MIQE 2.0 and the Urgent Need to Rethink qPCR Standards [https://www.mdpi.com/1422-0067/26/11/4975]
- 109. Quantitative PCR validation for research scientists: Part 2 of 2 [https://virologyresearchservices.com/2023/01/13/quantitative-pcr-validation-for-research-scientists-part-2-of-2/]