Toward Global Standards: A Comprehensive Framework for Standardizing Sperm Epigenetic Protocols in Clinical and Research Laboratories
The critical role of sperm epigenetics in male fertility, embryonic development, and transgenerational health is now undisputed.
Toward Global Standards: A Comprehensive Framework for Standardizing Sperm Epigenetic Protocols in Clinical and Research Laboratories
Abstract
The critical role of sperm epigenetics in male fertility, embryonic development, and transgenerational health is now undisputed. However, the translation of this knowledge into clinical practice is hampered by a lack of standardized laboratory protocols. This article addresses the urgent need for harmonization by exploring the foundational principles of sperm epigenetics, proposing robust methodological pipelines for DNA methylation and sncRNA analysis, outlining strategies for troubleshooting and quality control, and establishing frameworks for multi-center validation. By providing a detailed roadmap, this work aims to bridge the gap between cutting-edge research and reliable, reproducible clinical diagnostics, ultimately improving patient care and advancing the field of reproductive medicine.
The Sperm Epigenome: Decoding its Role in Fertility and Transgenerational Inheritance
FAQ: Core Concepts and Mechanisms
FAQ 1: What are the core epigenetic mechanisms regulating spermatogenesis? Spermatogenesis is precisely controlled by at least three key epigenetic mechanisms: DNA methylation, histone modifications, and small non-coding RNAs (sncRNAs). These mechanisms work synergistically to control gene expression without altering the DNA sequence, ensuring the successful development of spermatogonial stem cells into mature spermatozoa. Their proper function is critical for male fertility, and dysfunction is strongly linked to infertility [1] [2].
FAQ 2: How does DNA methylation dynamically change during spermatogenesis? DNA methylation undergoes waves of erasure and re-establishment. In mouse Primordial Germ Cells (PGCs), global DNA demethylation occurs around embryonic days 8.5 to 13.5, reducing 5mC levels to about 16.3%. De novo methylation is then re-established from E13.5 to birth. After birth, methylation levels generally increase during the transition from undifferentiated to differentiating spermatogonia, with some demethylation occurring in preleptotene spermatocytes before reaching high levels in pachytene spermatocytes [1].
FAQ 3: What are the primary functions of sperm-borne sncRNAs? Sperm-borne sncRNAs, including miRNAs, piRNAs, and tsRNAs, are no longer seen as mere byproducts. They are now recognized as crucial carriers of epigenetic information. Upon fertilization, they can influence early embryonic gene expression and are implicated in the transgenerational inheritance of paternally acquired traits, especially those influenced by environmental factors [3].
FAQ 4: Why is histone modification so important during spermiogenesis? Histone modifications are essential for the dramatic chromatin remodeling that occurs as round spermatids mature. Key modifications, such as the hyperacetylation of lysine residues on histone H4, facilitate the critical replacement of histones with protamines. This exchange is necessary for achieving the extreme nuclear compaction and DNA silencing required for producing functional sperm [4] [2].
Troubleshooting Common Experimental Challenges
Issue 1: Inconsistent DNA Methylation Results in Sperm Samples
- Problem: High variability in bisulfite sequencing data from patient sperm samples.
- Solution:
- Standardized Cell Purity: Ensure pure sperm cell populations by using rigorous somatic cell lysis protocols before DNA extraction. Contamination by somatic cells (e.g., white blood cells) with different methylomes is a major confounder.
- Controlled Bisulfite Conversion: Implement a quality control step post-conversion using control oligonucleotides with known methylation status to confirm complete and non-degradative conversion.
- Batch Effect Correction: Process cases and controls in the same experimental batch. Include internal control samples (e.g., a reference sperm DNA sample) in each batch to allow for technical normalization during bioinformatic analysis [5].
Issue 2: Low Yield of sncRNAs from Mature Sperm
- Problem: Difficulty in extracting sufficient quantity and quality of sncRNAs from mature spermatozoa due to highly compacted chromatin and low cytoplasmic content.
- Solution:
- Optimized Lysis: Use a lysis buffer containing strong denaturants and reducing agents to effectively disrupt the dense, disulfide-bonded sperm nucleus and release nucleoprotein-complexed RNAs.
- DNase Treatment: Perform rigorous on-column DNase digestion to remove the vast excess of genomic DNA, which can co-purify and interfere with downstream RNA sequencing library preparation.
- Spike-in Controls: Use synthetic RNA spike-ins (e.g., from other species) during the extraction process to monitor technical efficiency and potential bias in RNA recovery [3].
Issue 3: Failure to Detect Specific Histone Modifications in Testicular Tissue
- Problem: Weak or non-specific signals in Chromatin Immunoprecipitation (ChIP) assays for histone marks in testis samples.
- Solution:
- Antibody Validation: Use antibodies validated for ChIP-specificity in testicular tissue. Check cited literature for which antibodies have been successfully used in spermatogenic cells.
- Cross-linking Optimization: Titrate formaldehyde concentration and cross-linking time. Over-cross-linking can mask epitopes, especially in highly compacted sperm chromatin.
- Chromatin Shearing: Optimize sonication conditions to achieve fragments between 200-500 bp. The unique chromatin landscape in spermatogenic cells may require more intensive sonication than somatic cells [2].
Data Presentation: Key Epigenetic Regulators and Alterations in Male Infertility
Table 1: DNA Methylation/Demethylation Enzymes and Their Roles in Spermatogenesis
| Enzyme/Protein | Function | Consequence of Loss-of-Function in Models |
|---|---|---|
| DNMT1 | Maintenance methyltransferase | Apoptosis of germline stem cells; Hypogonadism and meiotic arrest [1] |
| DNMT3A | De novo methyltransferase | Abnormal spermatogonial function [1] |
| DNMT3C | De novo methyltransferase | Severe defect in DSB repair and homologous chromosome synapsis during meiosis [1] |
| TET1 | DNA demethylation | Fertile, but mice show a progressive decline in spermatogonia numbers [1] [4] |
Table 2: Genes with Aberrant Sperm DNA Methylation Linked to Male Infertility
| Condition | Gene Name | Methylation Status | Proposed Function |
|---|---|---|---|
| Oligo-/Astheno-/ Teratozoospermia | MEST | Hypermethylation | Hydrolase activity [4] |
| DAZL | Hypermethylation | Germ cell development and differentiation [4] | |
| H19 | Hypomethylation | Imprinted gene; affects sperm concentration and motility [4] | |
| GNAS | Hypomethylation | G-protein alpha subunit [4] | |
| Non-Obstructive Azoospermia (NOA) | SOX30 | Hypermethylation | Transcription factor critical for spermatogenesis [4] |
Experimental Protocols
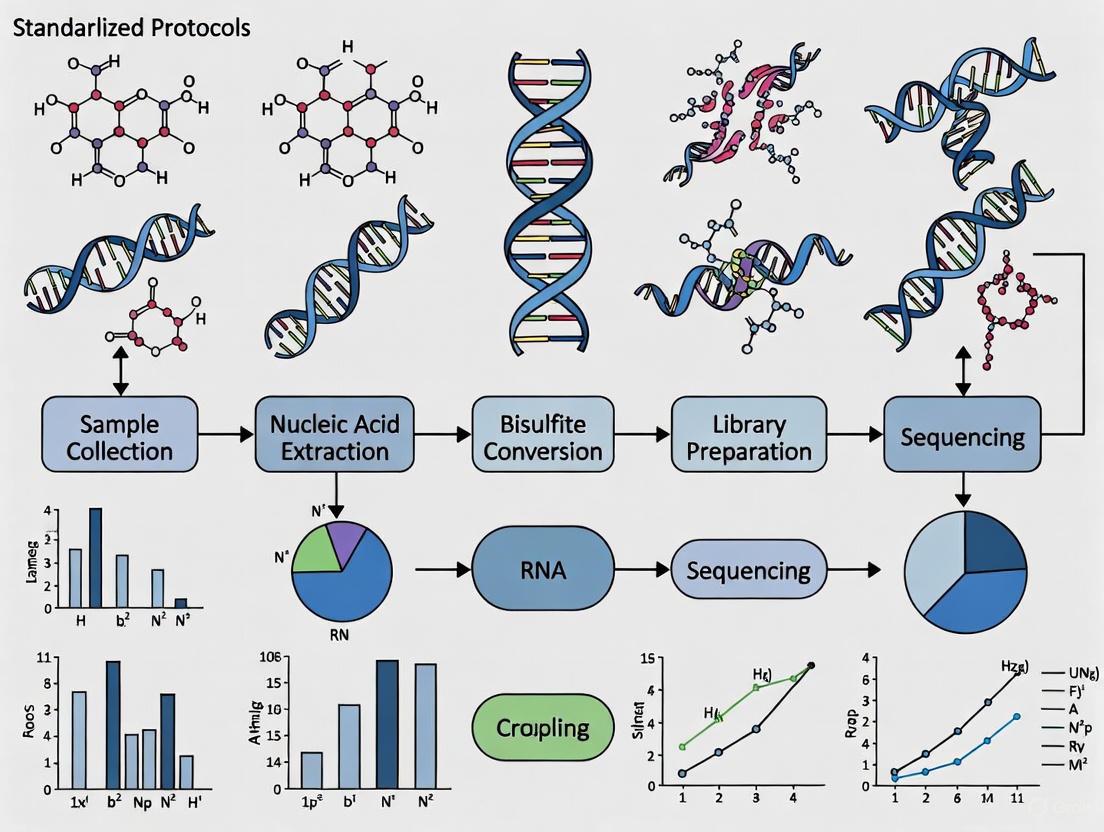
Protocol 1: Genome-wide DNA Methylation Analysis of Human Sperm using MeDIP-Seq This protocol is based on the method used to identify epigenetic biomarkers for male infertility and FSH therapy responsiveness [6].
- Sperm Collection and DNA Extraction: Collect semen samples after 2-5 days of sexual abstinence. Purify sperm cells using a discontinuous density gradient to remove somatic cells and debris. Extract genomic DNA using a column-based kit designed for sperm cells, which often have highly cross-linked chromatin.
- DNA Fragmentation and Quality Control: Fragment the purified DNA by sonication to an average size of 200-500 bp. Verify the fragment size distribution using a bioanalyzer.
- Methylated DNA Immunoprecipitation (MeDIP): Denature the fragmented DNA to generate single strands. Incubate with an antibody specific for 5-methylcytosine (5mC). Capture the antibody-DNA complexes using magnetic beads coated with protein A/G. Wash the beads stringently to remove non-specifically bound DNA.
- Elution and Library Preparation: Elute the immunoprecipitated methylated DNA from the beads. Prepare a next-generation sequencing library from this enriched DNA, as well as from an input DNA control (non-immunoprecipitated).
- Sequencing and Bioinformatic Analysis: Perform high-throughput sequencing (e.g., Illumina). Align sequences to the reference genome and use bioinformatic tools to identify Differential Methylated Regions (DMRs) between case and control samples.
Protocol 2: Investigating sncRNA Transfer via Epididymosomes This protocol helps study the post-testicular maturation of sperm sncRNA payload [3].
- Epididymosome Isolation: Dissect the caput and cauda regions of the epididymis from a model organism (e.g., mouse). Flush the luminal content or mince the tissue in a physiological buffer. Isolate epididymosomes by sequential ultracentrifugation (e.g., 10,000 x g to remove cell debris, followed by 100,000 x g to pellet vesicles).
- Sperm Collection: Collect testicular sperm from the seminiferous tubules (lacking epididymal modifications) and caput/cauda epididymal sperm.
- In Vitro Co-incubation: Incubate testicular sperm with isolated caput epididymosomes in a suitable culture medium for several hours at 34-37°C under 5% CO₂.
- RNA Extraction and Analysis: Extract total RNA from sperm before and after co-incubation. Analyze changes in the sncRNA profile using techniques like small RNA-Seq or RT-qPCR for specific sncRNAs (e.g., miRNAs, tsRNAs).
Signaling Pathway and Workflow Visualizations
Epigenetic Regulation of Spermatogenesis
Sperm sncRNA Acquisition Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Sperm Epigenetics Research
| Reagent/Category | Specific Examples | Critical Function in Research |
|---|---|---|
| DNA Methylation Analysis | Bisulfite Conversion Kit, Anti-5mC Antibody, DNMT/TET Antibodies | Converts unmethylated cytosines to uracils for sequencing; Enriches methylated DNA for MeDIP; Confirms protein expression and localization via WB/IHC. |
| Histone Modification Analysis | Antibodies for H3K4me3, H3K9me3, H3K27me3, Acetyl-H4 | Key for ChIP assays to map activating/repressive histone marks genome-wide in spermatogenic cells. |
| sncRNA Analysis | Small RNA-Seq Library Prep Kit, miRNA/tsRNA Inhibitors/Mimics | Enables profiling of sperm sncRNA populations; Used for functional validation of sncRNA roles in germ cells or early embryos. |
| Cell Isolation & Culture | Percoll/Density Gradient Media, Collagenase/DNase I, SSC Culture Media | Purifies viable sperm populations; Isolates testicular cells for primary culture; Supports in vitro self-renewal and differentiation of SSCs. |
| BMY-43748 | BMY-43748, MF:C20H17F3N4O3, MW:418.4 g/mol | Chemical Reagent |
| NCX899 | NCX899|NO-Releasing Enalapril Derivative | NCX899 is a nitric oxide (NO)-donating ACE inhibitor for hypertension research. This product is for Research Use Only (RUO). Not for human or veterinary use. |
Epigenetic dysregulation is increasingly recognized as a pivotal factor in male infertility, with DNA methylation, histone modifications, and chromatin remodeling playing critical roles in spermatogenesis and early embryonic development [1]. The standardization of experimental protocols across laboratories is essential for generating reproducible and clinically meaningful data. This technical support center provides troubleshooting guides, detailed methodologies, and reagent solutions to address the common challenges researchers face when investigating sperm epigenetics, thereby supporting the broader research objective of harmonizing analytical approaches in this rapidly advancing field.
Frequently Asked Questions (FAQs) on Sperm Epigenetics
1. What is the clinical evidence linking DNA methylation to male infertility? Comparative analyses of testicular biopsies from patients with non-obstructive azoospermia (NOA) have revealed differential expression profiles of DNA methyltransferases (DNMTs) compared to patients with normal spermatogenesis [1]. This dysfunction in the enzymes that establish and maintain DNA methylation patterns is strongly correlated with impaired spermatogenesis.
2. How does the sperm epigenome influence embryo development? Sperm delivers epigenetic instructions at fertilization that are required for the correct regulation of gene expression in the developing embryo. Key developmental genes in sperm lose activating marks (H3K4me2/3) and retain repressive marks (H3K27me3) during spermatid maturation. Experimental removal of these marks deregulates gene expression in the resulting embryos [7].
3. Can environmental factors alter the sperm epigenome? Yes, post-testicular oxidative stress can oxidize sperm DNA and is associated with significant changes in epigenetic marks, including an increase in overall DNA hydroxymethylation. Antioxidant supplementation can mitigate oxidative damage but may also induce mild epigenetic alterations, highlighting the need for careful clinical evaluation [8].
4. What is a "bivalent" chromatin domain, and why is it important? A bivalent chromatin domain contains both a repressive mark (H3K27me3) and an activating mark (H3K4me3) on the same promoter. In embryonic stem cells, this state poises key developmental regulators for either activation or silencing upon differentiation. A similar poising mechanism may be at work during spermatogenesis [9].
Troubleshooting Guides for Epigenetic Assays
DNA Methylation Analysis
Table 1: Troubleshooting DNA Methylation Experiments
| Problem Scenario | Expert Recommendation |
|---|---|
| Very little or no methylated DNA enrichment | When using low DNA input, strictly follow the protocol specified for that amount. MBD protein can bind non-methylated DNA to some extent if the protocol is not optimized [10]. |
| Particulate matter after adding bisulfite conversion reagent | Centrifuge the material at high speed and use only the clear supernatant for the conversion reaction [10]. |
| Poor amplification of bisulfite-converted DNA | Use primers 24-32 nts in length with no more than 2-3 mixed bases. Use a hot-start Taq polymerase (e.g., Platinum Taq) and aim for amplicons around 200 bp, as bisulfite treatment can cause strand breaks [10]. |
Chromatin Immunoprecipitation (ChIP)
A primary challenge in ChIP is antibody specificity and efficacy, which can vary widely [9]. Always validate antibodies for your specific application. Furthermore, the homogeneity of the starting cell population is critical for clear data interpretation [9]. When working with tissues like endometrium, stratifying results based on ChIP efficiency can reveal significant, region-specific findings that are otherwise masked [11].
Detailed Experimental Protocols
Protocol 1: Chromatin Immunoprecipitation (ChIP) from Tissue
This protocol is adapted from a 2025 study on endometrial tissue [11].
Workflow Overview:
Methodology:
- Tissue Preparation: Snap-freeze tissue in liquid nitrogen. Pulverize the frozen tissue using a mortar and pestle.
- Cross-linking: Dissolve ~50 µg of pulverized tissue in PBS with protease inhibitors. Homogenize with a Dounce homogenizer. Cross-link proteins to DNA with formaldehyde for 10 minutes, and quench the reaction with glycine.
- Chromatin Shearing: Isolate nuclei and shear DNA to 100–300 bp fragments using a sonicator (e.g., Bioruptor).
- Immunoprecipitation: Pre-clear chromatin with non-specific IgG and magnetic A/G beads. Divide the sample into three parts:
- Input (5%): Saved for normalization.
- Specific Antibody: Incubate with antibody against your target mark (e.g., H3K27me3).
- Control IgG: Incubate with non-specific IgG. Incubate samples overnight at 4°C with rotation. Add magnetic beads the next day and incubate for 3 hours.
- Washing and Elution: Wash beads sequentially with low-salt buffer, high-salt buffer, LiCl wash buffer, and TE buffer. Elute the chromatin from the beads.
- Decrosslinking and Purification: Treat the eluate with RNase A, NaCl, and proteinase K at 45°C for 2 hours to reverse cross-links. Purify the DNA using a commercial kit.
- Analysis: Quantify the enriched DNA by qPCR using primers for your regions of interest. Always include a positive control locus (e.g., MYT1 promoter) [11]. Normalize results to the input and IgG control signals.
Protocol 2: Assessing the Impact of Oxidative Stress on Sperm DNA (Mouse Model)
This protocol is adapted from a 2024 study investigating post-testicular oxidative stress [8].
Workflow Overview:
Methodology:
- Animal Model: Use transgenic mouse models susceptible to sperm DNA oxidation (e.g.,
Gpx5−/−or double knockoutsnGpx4−/−; Gpx5−/−mice). Wild-type mice serve as controls [8]. - Intervention: Administer oral antioxidant supplementation (e.g., Fertilix) via drinking water for 14 days. A control group receives no supplementation.
- Sperm Retrieval: Euthanize mice and remove the cauda epididymides. Puncture and squeeze the tissue in M2 medium. Allow sperm to disperse for 10 minutes at 37°C and count sperm using a hemocytometer.
- DNA Extraction: Centrifuge sperm and resuspend the pellet. Incubate with RNAse A, followed by Proteinase K and SDS/DTT solutions to digest RNA and proteins. Extract genomic DNA using standard phenol-chloroform methods or a commercial kit.
- Analysis:
- DNA Oxidation: Quantify the oxidized base 8-OHdG using techniques like slot blot or ELISA.
- Epigenetics: Analyze global DNA methylation (5mC) and hydroxymethylation (5hmC) levels via slot blot with specific antibodies or enzymatic assays.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Sperm Epigenetic Research
| Reagent / Kit | Function and Application |
|---|---|
| DNMT & TET Enzymes | Writers (DNMTs) and erasers (TETs) of DNA methylation. Critical for dynamic methylation changes during spermatogenesis [1]. |
| Histone Modification Antibodies | Highly specific antibodies (e.g., for H3K4me3, H3K27me3) are essential for ChIP assays to map histone modification landscapes [9] [11]. |
| MBD Proteins | Methyl-binding domain proteins used to enrich methylated DNA fragments from samples for downstream analysis [1] [10]. |
| Bisulfite Conversion Kits | Chemical treatment that converts unmethylated cytosines to uracils, allowing for the precise mapping of methylated cytosines via sequencing or PCR [10]. |
| Chromatin Immunoprecipitation Kits | Provide optimized buffers, beads, and protocols for efficient and specific enrichment of chromatin bound by specific proteins or histone marks [11]. |
| Platinum Taq DNA Polymerase | A hot-start polymerase recommended for robust amplification of bisulfite-converted DNA, which is notoriously difficult to PCR due to its low complexity [10]. |
| RP 70676 | RP 70676, MF:C25H28N4S, MW:416.6 g/mol |
| Rosmarinic Acid | Rosmarinic Acid|High-Purity Reference Standard |
Oxidative Stress as a Key Disruptor of Sperm Epigenetic Patterns
Male infertility is a significant global health concern, affecting approximately 8-12% of couples of childbearing age, with male factors contributing to nearly 50% of cases [12]. Oxidative stress, resulting from an imbalance between reactive oxygen species (ROS) production and antioxidant defenses, has emerged as a major contributor to sperm dysfunction [13] [14]. Beyond its well-documented effects on sperm motility and DNA integrity, oxidative stress disrupts the precise epigenetic programming required for normal spermatogenesis and embryogenesis [13] [15].
Sperm cells are particularly vulnerable to oxidative damage due to their high polyunsaturated fatty acid content in membranes, limited cytoplasmic volume, and minimal antioxidant defenses [12]. The epigenome of spermatozoa—comprising DNA methylation patterns, histone modifications, and non-coding RNA profiles—is highly susceptible to oxidative insult [13]. These epigenetic modifications can persist through fertilization and impact embryonic development, potentially contributing to transgenerational inheritance of disease susceptibility [13] [16].
Understanding the mechanisms by which oxidative stress disrupts sperm epigenetic patterns is crucial for developing standardized diagnostic and therapeutic approaches in clinical and research settings. This technical guide addresses common challenges and provides troubleshooting recommendations for researchers investigating this critical interface between oxidative stress and epigenetic regulation in male reproduction.
Core Mechanisms: How Oxidative Stress Disrupts Sperm Epigenetics
Molecular Pathways of Epigenetic Dysregulation
Excessive ROS directly targets all major epigenetic regulatory systems in spermatozoa through multiple interconnected mechanisms:
DNA Methylation Alterations: Oxidative stress induces both global hypomethylation and gene-specific hypermethylation by several mechanisms. ROS oxidize cysteine residues in DNA methyltransferases (DNMTs), impairing their catalytic activity and leading to aberrant methylation patterns [13]. Additionally, oxidative base lesions like 8-hydroxy-2'-deoxyguanosine (8-OHdG) interfere with DNMT binding, preventing proper maintenance of methylation patterns [12] [15]. The oxidation of methyl group donors such as S-adenosyl-methionine (SAM) further disrupts methylation reactions [15].
Histone Modification Disruptions: ROS alter the activity of histone-modifying enzymes including histone acetyltransferases (HATs), histone deacetylases (HDACs), and histone methyltransferases [13]. This leads to abnormal histone acetylation and methylation patterns that compromise chromatin remodeling during spermatogenesis [13] [17]. Oxidative conditions also promote histone citrullination via peptidylarginine deiminase (PAD) activation, particularly affecting histone H3 and contributing to chromatin decondensation [17].
Non-Coding RNA Dysregulation: Oxidative stress alters the expression profiles of sperm microRNAs (miRNAs) including miR-34c, miR-34b, and miR-122, which regulate critical processes such as apoptosis, sperm production, and germ cell survival [15]. These oxidative stress-induced miRNA alterations can be transmitted to the embryo during fertilization, potentially affecting early developmental programming [13] [15].
Table 1: Primary Epigenetic Alterations Induced by Oxidative Stress in Spermatozoa
| Epigenetic Mechanism | Specific Alterations | Functional Consequences |
|---|---|---|
| DNA Methylation | Global hypomethylation; Gene-specific hypermethylation (MTHFR, NTF3, IGF2, H19) | Altered genomic imprinting; Impaired spermatogenesis; Reduced embryonic viability |
| Histone Modifications | Abnormal acetylation/methylation patterns; Increased histone citrullination | Defective chromatin compaction; Disrupted protamine replacement |
| Non-coding RNA Expression | Altered miR-34c, miR-34b, miR-122, miR-449 profiles | Impaired sperm maturation; Dysregulated apoptosis; Compromised embryonic development |
Oxidative Stress Signaling and Epigenetic Disruption Pathways
The diagram below illustrates the interconnected pathways through which oxidative stress disrupts key epigenetic mechanisms in spermatozoa.
Troubleshooting Guide: Common Experimental Challenges and Solutions
Frequently Asked Questions
Q1: How do we distinguish between oxidative stress-induced epigenetic changes versus age-related epigenetic alterations in sperm samples?
Answer: This represents a significant methodological challenge requiring careful study design and data interpretation. Paternal age independently influences sperm epigenetics through clonal selection mechanisms in spermatogonial stem cells [18] [19]. Recent research using ultra-accurate DNA sequencing (NanoSeq) has identified 40 genes where mutations are positively selected during spermatogenesis, with prevalence increasing with age [18]. To distinguish these effects:
- Implement age-matched control groups in all experiments and perform stratified analysis
- Measure and document specific oxidative stress biomarkers (8-OHdG, lipid peroxidation products, antioxidant enzyme activities) alongside epigenetic analyses
- Account for clonal expansion events by analyzing mutation patterns in addition to epigenetic marks
- Use longitudinal study designs when possible to track changes within individuals over time
Q2: What are the most reliable biomarkers for assessing oxidative stress in sperm samples?
Answer: A multi-parameter approach is recommended for comprehensive assessment:
Table 2: Biomarkers for Assessing Oxidative Stress in Sperm Samples
| Biomarker Category | Specific Markers | Methodology | Technical Considerations |
|---|---|---|---|
| Direct ROS Measurement | Chemiluminescence assays | Luminol-based probes | Requires fresh samples; Susceptible to interference |
| Lipid Peroxidation | Malondialdehyde (MDA), 4-hydroxynonenal (4-HNE) | HPLC, ELISA, TBARS assay | Standardize sample processing to avoid ex vivo oxidation |
| DNA Oxidation | 8-hydroxy-2'-deoxyguanosine (8-OHdG) | HPLC-MS/MS, Immunofluorescence | Correlates with DNA fragmentation and poor pregnancy outcomes |
| Protein Oxidation | Protein carbonyls, nitrotyrosine | Western blot, ELISA | Indicates advanced oxidative damage |
| Antioxidant Capacity | Total antioxidant capacity (TAC) | Colorimetric assays | Assesses overall defense status |
Q3: Why do we observe inconsistent DNA methylation patterns across different sperm samples from the same patient under similar oxidative stress conditions?
Answer: This heterogeneity arises from several sources:
- Varied susceptibility of spermatogenic stages: Different developmental stages of germ cells exhibit varying sensitivity to oxidative stress
- Clonal expansion dynamics: Mutations conferring selective advantages lead to non-random distribution of epigenetic alterations across sperm populations [18]
- Epigenetic plasticity: The epigenome undergoes active remodeling throughout spermatogenesis, creating windows of varying vulnerability
- Technical considerations: Include batch effects in processing, storage conditions affecting epigenome stability, and regional heterogeneity in epigenetic patterns within semen samples
Standardization Recommendation: Process multiple aliquots from each sample, implement rigorous quality control for bisulfite conversion efficiency in methylation studies, and use internal reference standards to normalize technical variability.
Q4: How can we minimize ex vivo oxidative damage during sperm sample processing for epigenetic analysis?
Answer: Implement a comprehensive antioxidant strategy throughout processing:
- Collection: Use antioxidant-containing collection tubes and process samples within 30 minutes of ejaculation
- Processing: Work under reduced oxygen conditions (anaerobic chambers) when possible; Use media supplemented with combined antioxidants (vitamin C, vitamin E, N-acetylcysteine)
- Cryopreservation: Include cryoprotectants with antioxidant properties; Use controlled-rate freezing to minimize ice crystal formation
- Storage: Maintain consistent temperature control; Avoid repeated freeze-thaw cycles; Store in aliquots to minimize oxygen exposure
Experimental Protocols: Standardized Methodologies
Comprehensive Assessment of Oxidative Stress and Epigenetic Parameters
Workflow Overview: This integrated protocol enables simultaneous assessment of oxidative stress parameters and epigenetic marks from the same sperm sample, reducing inter-sample variability.
Step-by-Step Protocol:
Sample Collection and Initial Processing
- Collect semen samples after recommended 2-3 days of sexual abstinence
- Allow liquefaction for 20-30 minutes at 37°C
- Assess basic parameters (count, motility, morphology) according to WHO guidelines
- Perform sperm separation using density gradient centrifugation
- Aliquot samples for parallel oxidative stress and epigenetic analyses
Oxidative Stress Assessment
- ROS Measurement: Use chemiluminescence assay with luminol probe
- Incubate 1×10^6 sperm with 5μM luminol for 15 minutes
- Measure chemiluminescence in microplate luminometer
- Express results as relative light units (RLU)/10^6 sperm
- Lipid Peroxidation: Quantify MDA using thiobarbituric acid reactive substances (TBARS) assay
- Incubate sperm lysate with TBA reagent at 95°C for 60 minutes
- Measure fluorescence at 532nm excitation/553nm emission
- Calculate concentration using tetraethoxypropane standard curve
- DNA Oxidation: Quantify 8-OHdG by ELISA
- Extract DNA using commercial kits with antioxidant preservatives
- Use competitive ELISA with anti-8-OHdG antibody
- Express as 8-OHdG/10^5 deoxyguanosine
- ROS Measurement: Use chemiluminescence assay with luminol probe
Epigenetic Analysis
- DNA Methylation: Perform whole-genome bisulfite sequencing (WGBS)
- Extract high-molecular-weight DNA
- Treat with bisulfite using commercial kits with efficiency controls
- Sequence on appropriate platform (minimum 10X coverage)
- Analyze differentially methylated regions (DMRs) bioinformatically
- Histone Modifications: Conduct chromatin immunoprecipitation sequencing (ChIP-seq)
- Cross-link chromatin with 1% formaldehyde for 10 minutes
- Sonicate to 200-500bp fragments
- Immunoprecipitate with antibodies against specific histone marks (H3K4me3, H3K27ac, H3K9me3)
- Sequence immunoprecipitated DNA and input controls
- miRNA Profiling: Implement small RNA sequencing
- Extract total RNA including small fractions
- Construct libraries with adapters for small RNAs
- Sequence on appropriate platform
- Quantify known miRNAs and identify novel species
- DNA Methylation: Perform whole-genome bisulfite sequencing (WGBS)
Intervention Studies: Antioxidant Supplementation Protocol
Objective: To evaluate the efficacy of antioxidant interventions in reversing oxidative stress-induced epigenetic alterations.
Experimental Design:
- Duration: 3-month intervention period based on established spermatogenesis cycles
- Groups: Randomized, placebo-controlled design with pre- and post-intervention sampling
- Participants: Men with confirmed oxidative stress (elevated ROS and DNA fragmentation)
Antioxidant Formulation:
- Vitamin C: 1000mg/day
- Vitamin E: 800IU/day
- N-acetylcysteine: 600mg/day
- Coenzyme Q10: 200mg/day
- Zinc: 50mg/day
Assessment Timeline:
- Baseline assessment (epigenetic, oxidative stress, conventional semen parameters)
- Monthly follow-up for adherence and safety monitoring
- Endpoint assessment (3 months) repeating baseline measurements
- Optional extended follow-up for persistence assessment
Research Reagent Solutions: Essential Materials for Investigation
Table 3: Essential Research Reagents for Studying Oxidative Stress and Sperm Epigenetics
| Reagent Category | Specific Products | Application Notes |
|---|---|---|
| ROS Detection | Luminol, DCFDA, MitoSOX Red | MitoSOX specifically detects mitochondrial superoxide; Validate with positive controls |
| Oxidative Damage Kits | OxiSelect TBARS Assay, 8-OHdG ELISA | Include internal standards; Run in duplicate |
| DNA Methylation | EZ DNA Methylation kits, NEBNext Enzymatic Methyl-seq | Bisulfite conversion efficiency >99% required |
| Histone Modification | Active Motif ChIP kits, specific antibodies (H3K4me3, H3K27ac) | Verify antibody specificity with peptide blocks |
| miRNA Analysis | Qiagen miRNeasy, Illumina Small RNA Library Prep | Include spike-in controls for normalization |
| Antioxidants | N-acetylcysteine, Vitamin E (Trolox), MitoTEMPO | Use fresh preparations; Protect from light |
| Sperm Processing | SpermGrad, Human Tubal Fluid media | Use protein-supplemented media for processing |
Data Interpretation Guidelines: Standards for Analysis and Reporting
Quality Control Thresholds
Establish and report the following quality metrics for all experiments:
- Sample purity: >95% sperm cells in final analysis
- DNA integrity: DNA fragmentation index <30% for epigenetic studies
- Bisulfite conversion efficiency: >99% for methylation analyses
- Sequencing depth: Minimum 10X coverage for WGBS, 20 million reads for ChIP-seq
- Antioxidant validation: Include positive and negative controls in all oxidative stress assays
Statistical Considerations
- Account for multiple testing in epigenome-wide analyses (FDR <0.05)
- Include appropriate covariates in models (age, BMI, abstinence time)
- Report effect sizes alongside p-values for interventional studies
- For paired designs (pre-post intervention), use appropriate paired statistical tests
Standardizing methodologies for investigating oxidative stress-induced epigenetic disruptions in sperm is essential for generating comparable, reproducible data across laboratories. This technical guide provides a framework for comprehensive assessment, troubleshooting common challenges, and implementing rigorous experimental protocols. As research in this field advances, continued refinement of these standards will enhance our understanding of how oxidative stress compromises sperm epigenetic integrity and ultimately affects reproductive outcomes and intergenerational health.
For decades, the primary focus of developmental origins of health and disease has centered on maternal influences. However, a growing body of evidence demonstrates that paternal life experiences before conception significantly impact offspring health and development through epigenetic mechanisms [20] [21]. The paternal environment—including diet, stress, toxin exposure, and lifestyle choices—can induce epigenetic modifications in sperm that are transmitted to the embryo, potentially influencing metabolic function, neurodevelopment, and disease susceptibility in subsequent generations [22]. This technical support center addresses the critical need for standardized methodologies in the rapidly evolving field of paternal epigenetic inheritance research, providing troubleshooting guidance and experimental protocols to enhance reproducibility across laboratories.
Key Epigenetic Mechanisms in Paternal Inheritance
Sperm Epigenetic Landscape
Sperm possess a unique epigenome characterized by several distinct features that enable the transmission of paternal environmental information to the embryo. Key epigenetic marks in sperm include:
DNA Methylation: Cytosine methylation at CpG islands regulates gene expression and genomic imprinting [20]. Although most methylation marks are erased during embryonic reprogramming, some regions, particularly imprinted genes and transposable elements, can escape this process [23] [24].
Histone Modifications: During spermatogenesis, most histones are replaced by protamines to achieve highly compact chromatin [20]. However, approximately 5%-15% of histones are retained at specific genomic regions, particularly promoters of developmentally important genes [20] [7]. These histones carry modifications such as H3K4me2/3, H3K27me3, and H3K9me that can influence embryonic gene expression [7].
Non-coding RNAs: Sperm contain various non-coding RNAs, including microRNAs (miRNAs), tRNA-derived small RNAs (tsRNAs), and rRNA-derived small RNAs (rsRNAs) [22] [24]. These RNAs can directly influence embryonic development and mediate the transmission of paternal environmental exposures [22] [24].
Evading Embryonic Reprogramming
A significant challenge in understanding paternal epigenetic inheritance involves how sperm epigenetic marks evade the extensive reprogramming that occurs after fertilization. Research indicates that some epigenetic marks escape this reprogramming through:
- Erasure and Reestablishment: Some paternal epigenetic marks are erased during embryonic reprogramming but are subsequently reestablished during later developmental stages [24].
- Resistance to Erasure: Certain genomic regions, particularly imprinted genes and some transposable elements, demonstrate resistance to demethylation during reprogramming [23].
Studies investigating paternal stress exposure have shown that approximately 11.36% of differential DNA methylation regions (DMRs) in sperm demonstrate intergenerational inheritance, while 0.48% show transgenerational inheritance, persisting even to the F2 generation [24].
Experimental Models and Methodologies
Standardized Paternal Exposure Models
Several well-established experimental models are used to study paternal epigenetic inheritance:
Dietary Manipulation Models:
- High-Fat Diet (HFD): Typically 60% fat content administered for 8-12 weeks before mating [25]
- Low-Protein Diet: Usually 5-10% protein content for multiple generations [21]
- Methyl-Donor Supplementation: Folate, choline, betaine, and vitamin B12 [21]
Psychological Stress Models:
- Restraint Stress: Physical restriction for 2-6 hours daily over several weeks [24]
- Chronic Unpredictable Stress: Variable stressors applied unpredictably to prevent habituation [22]
Toxicant Exposure Models:
- Endocrine Disruptors: Bisphenol A, phthalates administered via drinking water [20] [22]
- Heavy Metals: Cadmium, lead exposure studies [22]
- Air Pollutants: PM2.5 exposure systems [22]
Quantitative Data on Paternal Exposure Effects
Table 1: Paternal Exposure Effects on Offspring Health Outcomes
| Paternal Exposure | Experimental Model | Offspring Phenotypes | Epigenetic Mechanisms |
|---|---|---|---|
| High-Fat Diet [21] [25] | Mouse (C57BL/6) | Glucose intolerance, insulin resistance, increased body weight, altered lipid metabolism | Sperm DNA methylation changes, altered sncRNA expression (miRNA, tsRNA), H3K4me3 alterations |
| Psychological Stress [24] | Mouse (restraint stress) | Depressive-like behaviors, metabolic disorders, reproductive deficits | Differential DNA methylation regions (DMRs), tsRNA and rsRNA dysregulation |
| Protein Restriction [21] | Mouse (low protein diet) | Impaired cardiovascular function, altered lipid metabolism, perturbed placental development | Modified DNA methylation, histone modifications, altered expression of imprinted genes |
| Endocrine Disruptors [20] [22] | Human cohorts & rodent models | Blastocyst quality reduction, reproductive abnormalities, metabolic changes | Altered DNA methylation in sperm, particularly at imprinted genes |
Table 2: Inheritance Patterns of Paternal Epigenetic Modifications
| Epigenetic Mark | Intergenerational Inheritance | Transgenerational Inheritance | Reprogramming Evasion Mechanism |
|---|---|---|---|
| DNA Methylation [24] | ~11.36% of stress-induced DMRs | ~0.48% of stress-induced DMRs | Erasure and subsequent reestablishment |
| Histone Modifications [7] | Retained at developmental promoters | Limited evidence | Protection during histone-to-protamine transition |
| tsRNAs/miRNAs [22] [24] | Altered expression in F1 | Limited evidence | Direct delivery to oocyte during fertilization |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Sperm Epigenetic Research
| Reagent/Category | Specific Examples | Research Application |
|---|---|---|
| Epigenetic Analysis Kits | Whole-genome bisulfite sequencing kits, ChIP-seq kits, MeDIP kits | Genome-wide methylation mapping, histone modification profiling |
| sncRNA Analysis Tools | Small RNA sequencing kits, tsRNA-enrichment protocols, miRNA inhibitors | sncRNA profiling and functional validation |
| Antibodies | Anti-5-methylcytosine, anti-H3K4me3, anti-H3K27me3, anti-H3K9me2 | Immunofluorescence, Western blot, chromatin immunoprecipitation |
| Sperm Processing Reagents | Protamine extraction buffers, histone purification kits, sperm lysis buffers | Sperm epigenetic mark isolation and analysis |
| Embryo Culture Media | KSOM, M16, specialized methyl donor-supplemented media | In vitro fertilization and preimplantation embryo culture |
| Necrostatin 2 racemate | Necrostatin 2 racemate, MF:C13H12ClN3O2, MW:277.70 g/mol | Chemical Reagent |
| Fluacrypyrim |
Troubleshooting Guides and FAQs
Common Experimental Challenges and Solutions
Q1: We observe high variability in offspring phenotypes despite controlled paternal exposures. What factors might contribute to this inconsistency?
A: Several factors can contribute to phenotypic variability:
- Strain-specific effects: C57BL/6 mice show different susceptibility to paternal diet effects compared to other strains [25]. Consistently use the same genetic background.
- Exposure timing and duration: The same stressor applied during different developmental windows (early life vs. adulthood) produces distinct epigenetic outcomes [20]. Standardize exposure protocols.
- Sperm heterogeneity: Use pooled sperm from multiple animals for epigenomic analyses to minimize individual variation.
- Maternal environment: Even when using control females, subtle variations in maternal care can influence offspring phenotypes. Cross-foster pups to control for this effect.
Q2: Our analyses detect only weak epigenetic signals in offspring tissues despite strong paternal exposure effects. How can we enhance detection sensitivity?
A: This common challenge arises because only a small proportion of paternal epigenetic marks evade reprogramming:
- Focus on resilient genomic regions: Prioritize analysis of imprinted genes, transposable elements, and metastable epialleles known to resist reprogramming [23] [24].
- Increase sequencing depth: For bisulfite sequencing, aim for >20X coverage to detect subtle methylation changes [24].
- Analyze multiple offspring: Pool data from multiple F1 individuals to identify consistently altered regions.
- Use targeted approaches: Instead of whole-genome analyses, focus on candidate regions identified in prior studies of similar exposures.
Q3: How can we distinguish true transgenerational inheritance from intergenerational effects?
A: Proper experimental design is critical:
- For paternal lineage studies: True transgenerational inheritance requires effects persisting in the F2 generation (great-grandchildren of exposed males), as the F1 generation (grandchildren) were directly exposed as germ cells within the F0 father [24].
- Include proper controls: Always compare against non-exposed control lineages maintained in parallel for the same number of generations.
- Track specific epigenetic marks: Use bisulfite sequencing or other epigenetic methods to confirm transmission of specific marks across generations [24].
Technical Methodology FAQs
Q4: What is the optimal method for isolating high-quality sperm for epigenetic analysis?
A: Standardized sperm isolation is crucial for reproducible results:
- Collection method: Use swim-up or density gradient centrifugation to isolate motile sperm and minimize somatic cell contamination.
- Processing time: Process samples within 2 hours of collection to prevent degradation of epigenetic marks.
- Storage conditions: Snap-freeze in liquid nitrogen and store at -80°C for DNA/RNA analyses; use cross-linking for histone studies.
- Quality assessment: Assess sperm viability, motility, and morphology before epigenetic analysis to control for sample quality.
Q5: Which epigenetic analysis platform provides the best balance between coverage and cost for sperm studies?
A: Platform selection depends on research goals and resources:
- Reduced representation bisulfite sequencing (RRBS): Cost-effective for CpG-rich regions, suitable for initial screening.
- Whole-genome bisulfite sequencing (WGBS): Comprehensive coverage but more expensive; recommended for discovery-phase studies [24].
- EPIC array: Targeted approach for human studies covering >850,000 CpG sites.
- Small RNA sequencing: Essential for sncRNA profiling; requires specialized library preparation protocols for tsRNAs [22].
Q6: How can we control for potential maternal contributions in paternal inheritance studies?
A: Several strategies can help control for maternal effects:
- Use virgin control females: Ensure females have no prior mating experience or pregnancies.
- Standardize maternal environment: House all females under identical conditions before and during mating.
- Cross-fostering: Transfer pups to unaffected foster mothers immediately after birth to control for postnatal maternal effects.
- In vitro fertilization: Use IVF to completely control the maternal contribution during conception [7].
Standardization and Future Directions
The field of paternal epigenetic inheritance requires rigorous standardization to enhance reproducibility. Key considerations include:
- Reporting standards: Clearly document exposure paradigms, animal housing conditions, sperm collection methods, and analysis parameters.
- Epigenetic controls: Include reference samples in epigenetic analyses to control for technical batch effects.
- Multidimensional analysis: Integrate data from multiple epigenetic layers (DNA methylation, histone modifications, and sncRNAs) rather than focusing on single mechanisms.
- Replication across models: Validate findings in multiple experimental systems and, when possible, human cohorts.
As research progresses, standardized protocols for sperm epigenetic analysis will be essential for understanding the mechanisms underlying paternal inheritance and developing potential interventions to mitigate adverse transgenerational health effects.
The Impact of Paternal Age and Environmental Exposures on Sperm Epigenetic Integrity
Sperm epigenetics refers to the molecular modifications on sperm DNA that regulate gene expression without altering the underlying DNA sequence. These modifications include DNA methylation, histone modifications, and the presence of small non-coding RNAs (sncRNAs) [26]. The sperm epigenome is established during germ cell development and maturation and is crucial for proper sperm function, fertilization, and embryonic development [26]. Unlike female eggs, which are formed before birth, sperm are produced continuously throughout a man's post-pubertal life, making the sperm epigenome uniquely susceptible to modification by age and environmental exposures [27].
Key Sperm Epigenetic Modifications and Their Functions
Table 1: Core Components of the Sperm Epigenome
| Epigenetic Mark | Primary Function in Sperm | Impact on Offspring Health |
|---|---|---|
| DNA Methylation | Gene regulation, genomic imprinting, transposon silencing [26]. | Altered patterns linked to impaired embryo development and diseases like Beckwith-Wiedemann syndrome [26]. |
| Histone Modification | Chromatin compaction during protamination; retention at developmental gene promoters [26]. | Post-translational modifications can prevent proper histone removal, affecting genome programming [26]. |
| Small Non-Coding RNAs | Post-transcriptional gene regulation; potential role in intergenerational communication [26]. | Associated with transmission of paternal stress responses and metabolic phenotypes [26]. |
Impact of Paternal Age on Sperm Epigenetics
Increasing paternal age is associated with measurable declines in conventional sperm quality and integrity. Studies involving large cohorts have demonstrated that advancing age correlates with decreased semen volume, sperm motility, and increased sperm DNA fragmentation [28].
Table 2: Quantitative Impact of Paternal Age on Sperm Parameters (Data from [28])
| Paternal Age Group | Semen Volume | Progressive Motility | Total Motility | Sperm DNA Fragmentation Index (DFI) |
|---|---|---|---|---|
| 20-24 years | Baseline | Baseline | Baseline | Lowest |
| 25-29 years | -- | -- | -- | -- |
| 30-34 years | -- | -- | -- | -- |
| 35-39 years | Significant decline | Significant decline | Significant decline | Increased |
| >40 years | Lowest | Lowest | Lowest | Highest |
Beyond DNA fragmentation, aging also affects the sperm epigenome. The Paternal Age Effect (PAE) describes the accumulation of de novo mutations and epigenetic changes over time, increasing the risk of certain genetic syndromes and complex disorders like schizophrenia and bipolar disorder in offspring [27]. While sperm banks often set donor age limits at 40, the specific age thresholds for significant risk remain less defined than those for maternal age [27].
Impact of Environmental Exposures and Lifestyle on Sperm Epigenetics
Paternal lifestyle and environmental factors before conception can significantly alter the sperm epigenome, potentially affecting offspring health via epigenetic inheritance [26]. Key exposures include:
- Smoking: Induces DNA hypermethylation in genes related to anti-oxidation and insulin resistance [26].
- Obesity and Diet: Associated with greater risks of metabolic dysfunction (e.g., high blood glucose, increased body weight) in offspring via epigenetic alterations in sperm [26].
- Chronic Stress: Linked to metabolic changes and increased risk of depressive-like behavior and sensitivity to stress in offspring [26].
- Endocrine-Disrupting Chemicals (EDCs): Paternal exposure is linked to transgenerational transmission of increased disease predisposition, including infertility, testicular disorders, and obesity [26].
- Alcohol Consumption: Impacts the success of Assisted Reproductive Technology (ART) and can modify the sperm epigenome [26].
These factors can cause epimutations—heritable changes in gene expression that do not involve changes to the underlying DNA sequence. Some of these altered epigenetic marks can escape the widespread reprogramming that occurs after fertilization, allowing for potential intergenerational or transgenerational inheritance [29] [26].
Essential Research Reagent Solutions
Table 3: Key Reagents for Sperm Epigenetic Research
| Reagent / Material | Primary Function | Example Application |
|---|---|---|
| Somatic Cell Lysis Buffer (SCLB) | Selectively lyses contaminating somatic cells in semen samples [30]. | Purification of sperm population for downstream epigenetic analysis (e.g., DNA methylation). |
| Infinium Methylation BeadChip | Genome-wide analysis of DNA methylation at specific CpG sites [30]. | Profiling methylome; identifying somatic contamination using specific CpG markers. |
| Antibodies (e.g., 5-mdC) | Immunodetection of specific epigenetic marks, like 5-methylcytosine [31]. | ELISA-based global methylation quantification; immunoprecipitation for sequencing. |
| Enzymes (DNMTs, TETs) | Catalyze DNA methylation (DNMTs) and active demethylation (TETs) [26]. | In vitro studies to manipulate or understand the establishment/removal of methylation. |
| Bisulfite Conversion Reagents | Chemical treatment that converts unmethylated cytosine to uracil, leaving methylated cytosine unchanged [31]. | Foundation for bisulfite sequencing to map DNA methylation at single-base resolution. |
| Protamine-Specific Stains | Assess the efficiency of histone-to-protamine exchange during spermiogenesis [26]. | Evaluation of sperm chromatin maturity and packaging quality. |
Standard Operating Procedures (SOPs) and Critical Protocols
Protocol: Sperm Purification and Somatic Cell Contamination Control
Objective: To obtain a highly pure sperm population for epigenetic analysis, free from somatic cell contamination that can skew results [30].
- Initial Inspection: Wash fresh semen sample twice with 1X PBS by centrifugation at 200 g for 15 min at 4°C. Inspect the sample under a microscope (e.g., 20X objective) to identify the level of somatic cell contamination and perform a sperm count.
- Somatic Cell Lysis: Incubate the washed sample with freshly prepared Somatic Cell Lysis Buffer (SCLB) (0.1% SDS, 0.5% Triton X-100 in ddH2O) for 30 min at 4°C [30].
- Post-Lysis Inspection: Re-examine the sample under a microscope. If somatic cells are still detected, pellet the sample by centrifugation and repeat the SCLB treatment.
- Final Pellet: After confirming the absence of somatic cells, pellet the purified sperm by centrifugation, followed by a final PBS wash.
- Quality Control Checkpoints:
- Microscopy: Visual confirmation of somatic cell removal.
- Biomarker Analysis: Analyze DNA methylation at predefined CpG sites known to be highly methylated in somatic cells but hypomethylated in sperm (e.g., the 9,564 CpG sites identified by [30]). This serves as a molecular confirmation of purity.
- Data Analysis Cut-off: During differential methylation analysis, apply a conservative cut-off (e.g., 15%) to filter out potential bias from residual, undetectable contamination [30].
Protocol: Assessing Sperm DNA Fragmentation (SDF)
Objective: To evaluate the level of DNA damage in a sperm sample, a key parameter of male fertility and gamete quality [32] [33].
Multiple tests are available, each with strengths and limitations. The choice of test should be guided by the clinical question and laboratory capabilities [33].
Table 4: Common Sperm DNA Fragmentation (SDF) Tests
| Test Name | Methodology Principle | Key Metrics | Advantages | Disadvantages |
|---|---|---|---|---|
| TUNEL Assay [33] | Labels DNA strand breaks with fluorescent dUTP via terminal transferase. | % of TUNEL-positive sperm. | Direct measurement; can use flow cytometry or microscopy; suitable for low sperm counts. | High inter-laboratory variability; relatively expensive. |
| SCSA [33] | Measures DNA denaturation susceptibility using acridine orange stain. | DNA Fragmentation Index (DFI); High DNA Stainability (HDS). | High reproducibility; analysis of large cell numbers; sample can be frozen. | Requires expensive flow cytometer; requires high sperm concentration. |
| Comet Assay [33] | Electrophoresis-based visualization of DNA fragments. | Tail length, intensity, and moment. | Sensitive; can distinguish single vs. double-strand breaks; affordable. | Protocol not standardized; low throughput; potential for subjective analysis. |
| SCD Assay [33] | Acid denaturation and removal of nuclear proteins to reveal halo patterns. | Halo size (large halo = low fragmentation). | Simple, quick, economical; no complex instruments needed. | Halo can be difficult to score; sperm tail is not preserved. |
Frequently Asked Questions (FAQs): Troubleshooting Common Experimental Issues
Q1: Our sperm DNA methylation data from oligozoospermic samples shows widespread hypermethylation. How can we be sure this is a real biological signal and not an artifact from somatic cell contamination?
A: Somatic cell contamination is a major confounder, especially in samples with low sperm counts [30]. Implement a multi-layered quality control strategy:
- Rigorous Wet-Lab Purification: Always include a Somatic Cell Lysis Buffer (SCLB) treatment step in your sample processing protocol and confirm its efficacy under a microscope [30].
- Molecular QC: After DNA extraction, probe a panel of pre-identified CpG sites that are hypermethylated in blood/somatic cells but hypomethylated in pure sperm. A high signal in these markers indicates persistent contamination [30].
- Statistical Safeguard: During data analysis, apply a stringent differential methylation cut-off (e.g., ≥15-20%). This helps filter out spurious signals arising from low-level contamination that evades visual detection [30].
Q2: Which Sperm DNA Fragmentation (SDF) test should I implement in my clinical laboratory?
A: There is no single "best" test; the choice depends on your clinical goals and resources [33].
- For high-throughput, standardized testing with high reproducibility, the SCSA is a strong choice, though it requires a flow cytometer [33].
- For a direct, measurable test that can be used on very low sperm counts (e.g., from testicular biopsies), the TUNEL assay is advantageous [33].
- For a cost-effective and relatively simple method, the SCD (Halo) test is widely used in andrology labs [33].
- Consider the predictive value you need: TUNEL and the alkaline comet assay directly measure DNA breaks and may show closer correlations with ART outcomes [33].
Q3: How long can we store sperm for research purposes before the epigenome is significantly compromised?
A: Emerging evidence indicates that even short-term storage can induce epigenetic changes. A study on common carp sperm showed that 14 days of in vitro storage led to reduced sperm motility, increased DNA fragmentation, and altered DNA methylation patterns in both the sperm and the resulting offspring [31]. These changes were associated with functional alterations in the progeny, including impaired cardiac performance [31]. While optimized storage media can maintain fertilization capacity, researchers should be aware that storage duration itself is an experimental variable that can influence the sperm epigenome. It is critical to standardize and minimize storage times across experimental groups.
Q4: To what extent do paternal lifestyle-induced epigenetic changes get erased after fertilization and actually impact the offspring?
A: This is an active area of research. While a global epigenetic reprogramming occurs after fertilization, some epigenetic marks, particularly at imprinted gene control regions and certain transposable elements, can escape this erasure [26]. Furthermore, sperm carry small non-coding RNAs and retained histones with specific modifications that can influence gene expression in the early embryo [26]. Paternal exposure to factors like obesity, stress, or toxins can alter these marks, and studies in animal models have shown these changes can be associated with metabolic and behavioral phenotypes in the next generation [26]. The inheritance is likely probabilistic and context-dependent, not absolute.
Building a Robust Pipeline: From Sample Collection to Data Generation
Standard Operating Procedures for Sperm Collection, Processing, and Storage
Pre-Collection Procedures and Patient Preparation
Abstinence Period: Maintain sexual abstinence for two to seven days before sample collection. Record the date of the last ejaculation, as this information is critical for interpreting concentration and motility results. An abstinence period of less than two days can lower sperm count, while more than seven days can increase the proportion of immobile or degraded sperm [34].
Lifestyle and Environmental Factors: Patients should limit or avoid alcohol, tobacco, and recreational drugs for several days to a week before collection, as these substances are linked to reduced sperm quality. It is also crucial to avoid heat exposure to the testicles (e.g., hot tubs, saunas, tight underwear) in the 48 to 72 hours before collection. Patients should postpone collection if they have a fever or active infection, as illness can temporarily alter semen parameters [34].
Sample Identification and Documentation: Before collection, ensure all necessary materials are present, including a sterile collection cup, labels, and required paperwork. Proper patient identification and accurate documentation are essential for traceability and preventing sample mix-ups [35].
Sample Collection Protocols
Collection Method: The complete ejaculate must be collected directly into the sterile container provided by the clinic or laboratory. Using any other container, such as household jars, is prohibited. The inside of the cup or lid should not be touched, as this can introduce contaminants. The initial fraction of the ejaculate often contains the highest sperm concentration, so collecting the full sample is critical for accurate analysis [34].
Collection Environment: For at-home collection, the sample should be produced in a clean, private environment to minimize contamination risk. Lubricants, soaps, or oils should not be used during collection unless a special sperm-safe product is supplied by the clinic [34].
Transport and Temperature Control: The sealed specimen must be maintained at room or body temperature (20–37°C) during transport. The sample should be delivered to the laboratory promptly; a time frame of 30 to 60 minutes from collection to delivery is recommended to maintain sperm motility and viability for analysis [34].
Semen Processing and Cryopreservation
Fundamentals of Cryopreservation
Cryopreservation is a routine technique in assisted reproduction to preserve male genetic material for decades. The process involves several key steps: the addition of cryoprotective agents (CPAs), cooling to storage temperature in liquid nitrogen (-196°C), thawing, and removal of the CPA. The primary goal is to achieve the highest post-thaw cell survival rate possible [36].
Table 1: Common Cryopreservation Techniques for Human Spermatozoa
| Technique | Cooling Rate | Key Principles | Notes |
|---|---|---|---|
| Programmable Slow Freezing | 0.5°C/min to 0°C/min | Controlled, gradual temperature drop allows for cellular dehydration, minimizing intracellular ice crystal formation [36]. | Most commonly used technique [36]. |
| Freezing on Liquid Nitrogen Vapors | Rapid, but uncontrolled | Sample is placed in the vapors above liquid nitrogen before immersion. Simpler than programmable freezing [36]. | A common rapid freezing method. |
| Vitrification (Ultrarapid Freezing) | Hundreds to thousands of °C/min | Extremely high cooling rates solidify the solution into a glass-like state without ice crystals [36]. | Not yet universally accepted as clinically relevant for sperm [36]. |
Cryoprotectants and Additives
Cryoprotective agents (CPAs) are essential to reduce cryodamage caused by freezing and thawing.
- Permeable CPAs: These include glycerol, DMSO, ethylene glycol, and 1,2-propanediol. They cross the cell membrane and facilitate water movement out of the cell, preventing the formation of lethal intracellular ice [36].
- Non-Permeable CPAs: These include sucrose, trehalose, and glucose. They increase the osmotic pressure outside the cell, drawing water out and promoting dehydration [36]. Antioxidants are sometimes added in clinical trials to mitigate oxidative stress and potentially improve post-thaw sperm quality [36].
Laboratory Standards and Staffing
Laboratories performing andrology procedures must adhere to strict standards. In the United States, laboratories performing quantitative semen analysis must comply with Clinical Laboratory Improvement Amendments (CLIA) regulations and are typically registered as high-complexity laboratories [35]. Accreditation by bodies such as the College of American Pathologists (CAP) or The Joint Commission (TJC) is required for embryology laboratories in clinics that are members of the Society for Assisted Reproductive Technology (SART) [35].
Personnel requirements are also specified, as summarized in the table below.
Table 2: Minimum Staff Requirements for an Embryology Laboratory (Adapted from ASRM Guidelines) [35]
| Title | Minimum Education | Minimum Experience | Continuing Education |
|---|---|---|---|
| Laboratory Supervisor | Bachelor's degree in a chemical, physical, or biological science | 4 years (with BS/BA) | 24 hours every 2 years |
| Senior Embryologist | Bachelor's degree in a chemical, physical, or biological science | 3 years | 24 hours every 2 years |
| Embryologist | Bachelor's degree in a chemical, physical, or biological science | 2 years | 24 hours every 2 years |
Potential Cryodamage and Molecular Impacts
The process of cryopreservation generates structural and molecular alterations in spermatozoa, known collectively as cryodamage. Injuries occur due to oxidative, temperature, and osmotic stress, leading to [36]:
- Decreases in sperm motility and viability.
- Reductions in mitochondrial activity.
- Increases in DNA fragmentation (DNA integrity damage).
- Changes in the fluidity and integrity of the plasma membrane.
The plasma membrane, rich in fatty acids, is the primary site of cryoinjury. Temperature stress during freezing and thawing causes irreversible changes to membrane lipids and proteins, leading to a loss of membrane fluidity and barrier function [36].
Epigenetic Considerations in Sperm Processing
Beyond genetic material, sperm carry epigenetic information that can influence embryonic development. The sperm epigenome includes chromatin modifications, DNA methylation, and non-coding RNAs [37]. During spermiogenesis, most histones are replaced by protamines to compact the DNA, but ~1% of histones in mice and up to 15% in humans are retained at specific genomic locations [37]. These retained histones often bear modifications like H3K4me3 and are enriched at promoters of genes critical for embryonic development [37].
Processing and cryopreservation can potentially impact these delicate epigenetic marks. Therefore, standardizing protocols is essential not only for preserving sperm motility and viability but also for maintaining the integrity of the paternal epigenetic template, which is programmed to regulate gene expression in the resulting embryo [7] [37].
Research Reagent Solutions
Table 3: Essential Reagents for Sperm Cryopreservation Research
| Reagent / Material | Function / Application | Examples / Notes |
|---|---|---|
| Permeable Cryoprotectants | Cross the cell membrane to depress freezing point and prevent intracellular ice formation [36]. | Glycerol, DMSO, Ethylene Glycol, 1,2-Propanediol. |
| Non-Permeable Cryoprotectants | Increase extracellular osmotic pressure, promoting cellular dehydration [36]. | Sucrose, Trehalose, Glucose. |
| Antioxidants | Mitigate oxidative stress during processing and freezing that can damage sperm membranes and DNA [36]. | Varied; often used in clinical trials. Specific compounds not listed in results. |
| Liquid Nitrogen | Provides ultra-low temperature environment (-196°C) for long-term storage of cryopreserved samples [36]. | Used for storage and as a coolant in vapor freezing methods. |
| Sterile Collection Cups | Ensure aseptic collection of semen sample to prevent microbial contamination [34]. | Must be provided by the clinic/lab; household containers are not acceptable. |
Troubleshooting Guides and FAQs
Q1: What are the most common causes of poor post-thaw sperm motility, and how can they be addressed?
- Cause: Improper cooling rates during freezing. Rates that are too fast lead to intracellular ice formation, while rates that are too slow cause excessive dehydration and osmotic stress [36].
- Solution: Ensure the cryopreservation equipment is properly calibrated and that standardized protocols for slow freezing or vitrification are strictly followed. Optimizing the type and concentration of cryoprotectants can also help [36].
Q2: We are seeing high levels of DNA fragmentation in post-thaw samples. What could be the source of this?
- Cause: Cryopreservation-induced oxidative stress is a major factor leading to DNA damage [36].
- Solution: Consider the addition of antioxidants to the cryopreservation medium. Furthermore, review the processing steps to minimize physical and oxidative stress before the freezing procedure begins.
Q3: A patient's at-home collected sample arrived at the lab after 90 minutes. What is the impact, and should we process it?
- Impact: Sperm motility and viability progressively decline after collection. A 90-minute delay likely significantly reduces motility, compromising the analysis or usability for assisted reproductive technologies [34].
- Action: Process the sample but document the delay thoroughly on the report. The results should be interpreted with caution. For clinical use, discuss the potential impact with the physician and patient. Emphasize the importance of timely delivery (within 30-60 minutes) for future collections [34].
Q4: Why is standardizing the abstinence period so critical for research on sperm epigenetics?
- Rationale: The epigenetic landscape of sperm, including histone retention and DNA methylation, can be dynamic. Varying abstinence periods may introduce uncontrolled variability in these epigenetic marks, confounding research results. Standardization ensures that observed epigenetic differences are due to experimental conditions and not pre-analytical variables [34] [37].
Q5: Our laboratory is setting up a new sperm biobank for a large cohort study. What are the key considerations for ensuring sample quality and epigenetic integrity?
- Standardization: Implement and document identical SOPs for collection, processing, and freezing across all samples. This includes strict adherence to abstinence guidelines, consistent use of reagents and cryoprotectants, and uniform freezing protocols [35].
- Training & Competency: Ensure all staff are trained and competency-assessed according to guidelines (e.g., ASRM), performing a sufficient number of procedures annually to maintain proficiency [35].
- Storage & Monitoring: Use a robust inventory system with 24/7 temperature monitoring for liquid nitrogen storage tanks to ensure sample security and traceability [35].
The standardization of sperm epigenetic protocols across laboratories represents a critical step forward in male fertility research and assisted reproductive technology (ART). DNA methylation, a fundamental epigenetic mechanism involving the addition of a methyl group to cytosine bases, plays a crucial role in gene regulation, genomic imprinting, and embryonic development. For researchers and drug development professionals working in reproductive medicine, selecting the appropriate methylation profiling method is paramount for obtaining accurate, reproducible, and biologically relevant data. This technical support center provides a comprehensive comparison of three principal DNA methylation analysis techniques—Whole-Genome Bisulfite Sequencing (WGBS), Reduced Representation Bisulfite Sequencing (RRBS), and Methylation Microarrays—with a specific focus on their application in sperm epigenetics research. The following sections offer detailed troubleshooting guides, methodological protocols, and comparative frameworks to support experimental standardization across different laboratory settings.
Technical Comparison of Major DNA Methylation Assays
Whole-Genome Bisulfite Sequencing (WGBS) is considered the gold standard for DNA methylation analysis, providing single-base resolution methylation measurements across the entire genome. The method relies on sodium bisulfite conversion, which transforms unmethylated cytosines to uracils while leaving methylated cytosines unchanged, followed by next-generation sequencing [38]. This technique covers approximately 80% of all CpG sites in the human genome, enabling comprehensive methylation profiling [39].
Reduced Representation Bisulfite Sequencing (RRBS) offers a cost-effective alternative that combines restriction enzyme digestion with bisulfite sequencing. This method uses methylation-insensitive restriction enzymes (typically MspI) to digest genomic DNA, enriching for CpG-rich regions including promoters, CpG islands, and other regulatory elements. Following digestion, fragments are size-selected, bisulfite-treated, and sequenced [38]. While RRBS covers only about 5-10% of CpGs genome-wide, it focuses on functionally relevant regions with high CpG density [38].
Methylation Microarrays, particularly Illumina's Infinium platforms (such as the EPIC array), utilize bisulfite-converted DNA hybridized to oligonucleotide probes fixed on beads. The current EPIC array covers over 935,000 CpG sites, extensively profiling promoter regions, gene bodies, enhancers, and other regulatory elements [39] [40]. This technology provides a balanced approach between coverage, cost, and throughput, making it suitable for large-scale epigenetic studies.
Comparative Performance Table
Table 1: Technical comparison of WGBS, RRBS, and Methylation Microarrays for DNA methylation analysis
| Parameter | WGBS | RRBS | Methylation Microarrays |
|---|---|---|---|
| Resolution | Single-base | Single-base | Single-CpG (predefined sites) |
| Genomic Coverage | ~80% of CpGs (virtually entire genome) | ~5-10% of CpGs (CpG-rich regions) | >935,000 predefined CpG sites (EPIC array) |
| Coverage Bias | Minimal bias | Biased toward CpG islands and promoters | Designed coverage of regulatory regions |
| DNA Input Requirements | High (≥1μg) | Moderate (100-500ng) | Low (100-500ng) |
| DNA Degradation Concerns | High (bisulfite causes fragmentation) | High (bisulfite causes fragmentation) | Tolerant of partially degraded DNA, compatible with FFPE samples |
| Cost per Sample | High | Moderate | Low to moderate |
| Throughput | Low to moderate | Moderate | High |
| Data Analysis Complexity | High (requires advanced bioinformatics) | Moderate to high | Moderate (established pipelines) |
| Best Applications | Discovery-phase studies, novel biomarker identification, comprehensive methylation profiling | Cost-effective targeted methylation analysis, large cohort studies focusing on regulatory regions | Large-scale epidemiological studies, clinical biomarker validation, multi-center studies |
Emerging Alternative Technologies
Recent methodological advances have introduced new approaches that address limitations of traditional bisulfite-based methods. Enzymatic Methyl-Sequencing (EM-seq) utilizes enzymatic conversion rather than chemical bisulfite treatment, offering improved DNA preservation and reduced sequencing bias while maintaining single-base resolution [41] [38]. Oxford Nanopore Technologies (ONT) enables direct detection of methylation without conversion through long-read sequencing, providing access to challenging genomic regions and haplotype-resolution methylation profiling [41] [38]. These emerging technologies show particular promise for sperm epigenetics, where DNA integrity and comprehensive region coverage are paramount concerns.
Troubleshooting Guides and FAQs
Method Selection Guidance
Q: Which DNA methylation method is most suitable for establishing standardized sperm epigenetic protocols across multiple laboratories?
A: The choice depends on your specific research objectives, budget, and technical capabilities:
- For discovery-phase studies aiming to identify novel sperm-specific methylation biomarkers, WGBS provides the most comprehensive coverage [41] [39].
- For large-scale multi-center studies with hundreds to thousands of samples, methylation microarrays offer the best combination of throughput, reproducibility, and cost-effectiveness [40].
- For focused analysis of promoter regions and CpG islands with limited budget, RRBS represents a balanced option [38].
- For labs concerned about DNA degradation during bisulfite treatment, EM-seq provides a robust alternative with better DNA preservation [41] [38].
Consider starting with a pilot study comparing methods on a subset of samples to determine which approach best addresses your specific research questions before scaling up to full multi-center standardization.
Q: What method is most appropriate for analyzing limited sperm samples with low DNA concentration?
A: For limited sperm samples:
- Methylation microarrays typically require only 100-500ng of DNA and demonstrate good performance with suboptimal samples [40].
- RRBS can work with 100-500ng input, though lower inputs may affect coverage [38].
- WGBS generally requires ≥1μg of high-quality DNA, making it less suitable for limited samples [39].
- EM-seq shows improved performance with low-input samples compared to WGBS, potentially offering a good compromise between data quality and input requirements [38].
Technical Issue Resolution
Q: How can we address incomplete bisulfite conversion in our sperm DNA samples?
A: Incomplete bisulfite conversion leads to false positive methylation calls. To troubleshoot:
- Verify conversion efficiency by including synthetic unmethylated controls or assessing methylation levels at known unmethylated regions.
- Optimize conversion conditions by ensuring complete DNA denaturation before bisulfite treatment and preventing renaturation during conversion.
- Consider alternative methods such as EM-seq, which uses enzymatic conversion and may provide more consistent results, particularly for GC-rich regions like CpG islands [39] [38].
- Use fresh bisulfite reagents and strictly control reaction temperature and duration according to manufacturer recommendations.
Q: Our sperm samples show significant DNA fragmentation. How does this impact method selection?
A: DNA fragmentation presents particular challenges:
- WGBS and RRBS are highly sensitive to DNA fragmentation since bisulfite treatment further degrades DNA [39] [38].
- Methylation microarrays are more tolerant of partially degraded DNA and are compatible with FFPE samples, making them more robust for compromised sperm samples [40] [38].
- EM-seq causes less DNA damage than bisulfite treatment, offering advantages for fragmented samples [41] [38].
- Library preparation protocols specifically designed for degraded DNA may improve results regardless of the selected method.
Data Quality and Analysis
Q: What quality control metrics should we implement for cross-laboratory standardization?
A: Implement a comprehensive QC framework including:
- Bisulfite conversion efficiency (>99% recommended) for conversion-based methods [31].
- Coverage depth (≥10X for WGBS, ≥30X for RRBS) to ensure statistical power for methylation calling.
- Sample clustering to identify batch effects and outliers before integrative analysis.
- Positive controls including fully methylated and unmethylated standards across all batches.
- Reproducibility assessment through technical replicates across participating laboratories.
Q: How do we handle batch effects when combining data from multiple centers?
A: Batch effects are a major challenge in multi-center studies:
- Implement pre-harmonization protocols using standardized reagents, equipment, and procedures across centers.
- Include reference samples in each batch to monitor and correct for technical variation.
- Apply bioinformatic normalization such as Beta-mixture Quantile normalization for microarray data [39] [40].
- Design studies strategically by balancing experimental groups within each batch and center.
Experimental Workflows and Visualization
WGBS Experimental Workflow
Diagram 1: WGBS workflow showing key experimental steps.
The WGBS protocol begins with high-quality DNA extraction, requiring special attention to preserve DNA integrity. The critical bisulfite conversion step follows, where unmethylated cytosines are converted to uracils while methylated cytosines remain protected. This conversion enables discrimination between methylation states during sequencing. Library preparation incorporates adapters compatible with your sequencing platform, followed by deep sequencing to achieve sufficient coverage across the genome. Bioinformatics processing includes quality control, alignment to a bisulfite-converted reference genome, and methylation calling at individual cytosine positions [39] [38].
RRBS Experimental Workflow
Diagram 2: RRBS workflow with restriction digest and size selection.
The RRBS method begins with restriction enzyme digestion using MspI, which cuts at CCGG sites regardless of methylation status. Size selection enriches for fragments containing CpG-rich regions, significantly reducing the genomic space requiring sequencing. Following bisulfite conversion and library preparation, sequencing depth requirements are substantially lower than WGBS while maintaining single-base resolution in functionally relevant genomic regions [38] [42].
Microarray Experimental Workflow
Diagram 3: Microarray workflow with hybridization and scanning steps.
Methylation microarray analysis incorporates bisulfite conversion followed by whole-genome amplification to generate sufficient material for hybridization. The converted DNA is applied to the array, where it binds to locus-specific probes. Fluorescent detection determines the methylation status at each CpG site, with results typically reported as β-values ranging from 0 (completely unmethylated) to 1 (completely methylated) [39] [40].
The Scientist's Toolkit: Essential Research Reagents and Materials
Core Reagents for DNA Methylation Analysis
Table 2: Essential research reagents for DNA methylation analysis protocols
| Reagent/Material | Function | Method Compatibility | Technical Notes |
|---|---|---|---|
| Sodium Bisulfite | Chemical conversion of unmethylated cytosines to uracils | WGBS, RRBS, Microarrays | Critical to control conversion efficiency; causes DNA degradation |
| MspI Restriction Enzyme | Digests DNA at CCGG sites for reduced representation | RRBS | Methylation-independent cutting enables unbiased representation |
| DNA Methyltransferase Inhibitors | Prevents de novo methylation during sample processing | All methods | Particularly important for sperm samples with active enzymes |
| Bisulfite Conversion Kits | Optimized reagents for efficient cytosine conversion | WGBS, RRBS, Microarrays | Commercial kits improve reproducibility across laboratories |
| Methylated/Unmethylated Control DNA | Quality assessment of conversion efficiency | All methods | Essential for standardization across multiple laboratories |
| DNA Integrity Assessment Kits | Evaluates sample quality before processing | All methods | Critical for sperm samples which may have fragmentation issues |
| Bisulfite-Compatible Library Prep Kits | Preparation of sequencing libraries from converted DNA | WGBS, RRBS | Specialized kits account for bisulfite-induced sequence complexity reduction |
| Infinium MethylationEPIC Kit | Microarray-based methylation profiling | Microarrays | Covers >935,000 CpG sites including sperm-relevant regulatory regions |
| AA41612 | AA41612, MF:C12H15Cl2NO3S, MW:324.2 g/mol | Chemical Reagent | Bench Chemicals |
| GlyRS-IN-1 | GlyRS-IN-1, MF:C12H17N7O7S, MW:403.37 g/mol | Chemical Reagent | Bench Chemicals |
Standardization Considerations for Sperm Epigenetic Protocols
Sperm-Specific Methodological Challenges
Sperm cells present unique challenges for DNA methylation analysis due to their compact chromatin structure, high protamine content, and potential for DNA fragmentation. During standardizing sperm epigenetic protocols across laboratories, consider these critical factors:
DNA Extraction Optimization: Sperm DNA requires specialized extraction methods to efficiently break down disulfide bonds in protamines. Inconsistent extraction across laboratories can introduce significant variability in downstream methylation measurements. Implement a standardized protocol with rigorous quality control for DNA purity, concentration, and integrity.
Cell Purity Assessment: Sperm samples must be evaluated for contamination with somatic cells (especially white blood cells) which have distinct methylation patterns. Implement quality control measures such as microscopy or flow cytometry to ensure sample purity, as somatic cell contamination can dramatically alter perceived sperm methylation patterns.
Bisulfite Conversion Optimization: The highly compact nature of sperm chromatin may necessitate modified bisulfite conversion conditions to ensure complete denaturation and conversion. Consider extending denaturation times or using specialized denaturation buffers specifically validated for sperm DNA.
Multi-Center Standardization Framework
Establishing reproducible sperm methylation protocols across multiple laboratories requires a comprehensive standardization framework:
Reference Materials: Develop and distribute common reference sperm samples across participating laboratories to assess inter-lab reproducibility and enable data harmonization.
Standard Operating Procedures (SOPs): Create detailed, step-by-step protocols covering every aspect from sample collection through data analysis, with particular attention to critical steps that introduce technical variability.
Data Analysis Harmonization: Implement consistent bioinformatic pipelines for quality control, normalization, and methylation calling. For microarray data, apply standardized normalization methods like Beta-mixture Quantile normalization [39]. For sequencing-based methods, establish consistent alignment parameters, coverage thresholds, and methylation calling algorithms.
Proficiency Testing: Regularly assess laboratory performance through inter-laboratory comparisons and implement corrective actions when variability exceeds acceptable thresholds.
The comparative analysis of WGBS, RRBS, and methylation microarrays reveals distinct advantages and limitations for each method in the context of standardizing sperm epigenetic protocols. WGBS provides unparalleled comprehensive coverage but at higher cost and computational burden. RRBS offers a cost-effective alternative focused on functionally relevant regions. Methylation microarrays deliver high throughput and reproducibility ideal for multi-center studies. Emerging technologies like EM-seq and Oxford Nanopore sequencing present promising alternatives that may address certain limitations of established methods.
For laboratories embarking on sperm epigenetics standardization, the selection of methodology must align with specific research objectives, sample characteristics, and resource constraints. A phased approach—beginning with method validation using shared reference samples—provides the strongest foundation for successful multi-center implementation. As sperm epigenetic analysis continues to evolve, ongoing method refinement and standardization will be essential for advancing our understanding of male fertility and improving clinical outcomes in reproductive medicine.
Standardizing small non-coding RNA (sncRNA) profiling is critical for advancing research in male infertility. The distinct epigenetic landscape of sperm, rich in sncRNAs like microRNAs (miRNAs), transfer RNA-derived small RNAs (tsRNAs), and ribosomal RNA-derived small RNAs (rsRNAs), influences embryonic development and assisted reproductive technology outcomes [43]. Traditional RNA sequencing methods often fail to capture the full sncRNA spectrum due to technical biases, obscuring vital epigenetic information [44]. This guide provides standardized, troubleshooting-focused protocols to ensure comprehensive and reproducible sncRNA data across laboratories.
Frequently Asked Questions (FAQs) & Troubleshooting
1. Our small RNA-seq results show a sharp peak at ~22nt (miRNAs) but fail to detect longer sncRNAs (30-40nt) that are visible on the PAGE gel. What is the cause and how can we resolve this?
- Problem: This indicates a ligation and reverse transcription bias caused by RNA modifications. sncRNAs like tsRNAs and rsRNAs frequently possess terminal (e.g., 2',3'-cyclic phosphate) and internal (e.g., m1A, m22G) modifications that block adapter ligation and reverse transcription, making them undetectable in standard sequencing protocols [44] [45].
- Solution: Implement an enzymatic pre-treatment protocol to remove these modifications.
- Troubleshooting Tip: Always run a PAGE gel to visualize the total small RNA population before and after enzymatic treatment. If the post-treatment gel shows a richer smear in the 30-45nt range, your treatment was successful.
2. Why is the sncRNA profile from spermatozoa important for male infertility research, and which sncRNAs should we focus on?
- Answer: The sperm sncRNA profile is a key epigenetic regulator. Alterations in these profiles are linked to spermatogenic impairments, poor embryo development, and clinical outcomes of Assisted Reproductive Technology (ART) [43]. Beyond miRNAs, tsRNAs have emerged as crucial players. Studies using advanced profiling like PANDORA-seq have revealed that heat stress, a model for environmental infertility, causes significant and dynamic changes in tsRNAs, rsRNAs, and other sncRNAs in testicular tissue [45].
- Troubleshooting Tip: Do not limit your analysis to miRNAs. Ensure your bioinformatic pipeline is configured to annotate tsRNAs (derived from both pre-tRNA and mature tRNA), rsRNAs (from various ribosomal RNA subunits), and other noncanonical sncRNAs [45].
3. What is the optimal RNA size selection range for capturing a comprehensive sncRNA profile?
- Problem: Using a size selection cutoff of <30 nt, common in miRNA-focused protocols, will systematically exclude many tsRNAs and piRNAs [44].
- Solution: Adjust the size selection range to 15-45 nucleotides during library preparation to ensure the capture of miRNAs (~22nt), piRNAs (21-35nt), and the majority of tsRNAs and rsRNAs (30-40nt) [44] [45].
Methodological Deep Dive: Overcoming Technical Biases
The table below summarizes the core enzymatic treatments required to overcome key technical hurdles in sncRNA sequencing.
Table 1: Key Enzymatic Treatments for Unbiased sncRNA Profiling
| Enzyme | Primary Function | Problem Solved | Key Application Note |
|---|---|---|---|
| T4 Polynucleotide Kinase (T4 PNK) | Converts 2',3'-cyclic phosphates to 3'-phosphate/2'-OH ends; phosphorylates 5'-OH ends [45]. | Enables adapter ligation to sncRNAs with blocked termini, a common feature of tsRNAs and rsRNAs [44]. | Essential for capturing tsRNAs generated by cleavage via specific ribonucleases like ANG [46]. |
| AlkB Homolog (AlkB) | α-ketoglutarate-dependent dioxygenase that demethylates common internal modifications (e.g., m1A, m3C) [44] [45]. | Removes reverse transcription (RT)-blocking modifications, allowing cDNA synthesis to proceed through modified bases [44]. | Critical for the accurate quantification of a wide array of modified sncRNAs, dramatically altering the observed sncRNA landscape [45]. |
The following workflow diagram illustrates how these enzymatic steps are integrated into a standard library preparation protocol, known as PANDORA-seq.
Research Reagent Solutions
The table below lists essential reagents and kits used in the cited studies for robust sncRNA profiling.
Table 2: Essential Reagents for Advanced sncRNA Profiling
| Reagent / Kit | Function | Specific Example / Catalog Number |
|---|---|---|
| Specialized Library Prep Kit | Construction of sequencing libraries from small RNAs. | QIAseq miRNA Library Kit (QIAGEN: 331505) [45]. |
| Enzymatic Treatment Reagents | Key enzymes and buffers for removing RNA modifications. | AlkB (Epibiotek), T4 PNK (NEB: M0201L), PNK Buffer (NEB: B0201S), ATP (NEB: P0756S) [45]. |
| Reverse Transcription & qPCR Kits | Validation of sncRNA expression via RT-qPCR. | miRNA 1st Strand cDNA Synthesis Kit (Vazyme: MR101-01/02), miRNA Universal SYBR qPCR Master Mix (Vazyme: MQ101-01) [47]. |
| RNA Extraction Reagent | High-quality total RNA isolation. | TRIzol Reagent (Invitrogen; 15596018) [45]. |
Quantitative Data from Key Studies
The following table quantifies the dramatic impact of using bias-removing protocols on sncRNA discovery, which is fundamental for standardizing results across labs.
Table 3: Impact of PANDORA-seq on sncRNA Profile in Mouse Testis
| sncRNA Category | Standard RNA-Seq (Read %) | PANDORA-seq (Read %) | Fold-Change | Biological Relevance |
|---|---|---|---|---|
| miRNAs | ~31% | ~15% | ~2x decrease | Well-characterized in gene regulation; remains a core component. |
| tsRNAs | ~13% | ~43% | ~3.3x increase | Crucial in stress response (e.g., heat stress), spermatogenesis [45]. |
| rsRNAs | ~1% | ~7% | ~7x increase | Abundant but previously under-detected; potential novel biomarkers [44] [45]. |
| piRNAs | ~50% | ~29% | ~1.7x decrease | Still a major player, but previous abundance was overestimated due to missing other sncRNAs [45]. |
Data adapted from Shi et al. and related PANDORA-seq studies [44] [45].
Defining Quality Control Benchmarks for Bisulfite Conversion Efficiency and Library Preparation
In the field of sperm epigenetics, standardizing DNA methylation analysis across laboratories presents significant challenges, particularly in achieving consistent bisulfite conversion efficiency and reliable library preparation. Bisulfite conversion (BC) has served as the gold standard for DNA methylation profiling for decades, but this chemical process introduces substantial DNA fragmentation and loss, especially problematic with limited sperm samples. Recent advancements include enzymatic conversion (EC) methods that offer a gentler alternative, though with different performance characteristics. This technical support center provides troubleshooting guides and FAQs to address specific experimental issues, framed within the broader context of standardizing sperm epigenetic protocols across research laboratories. Establishing robust quality control benchmarks is essential for generating reproducible, high-quality data in male fertility research and diagnostic development.
Quantitative Performance Benchmarks
The performance of DNA conversion methods can be evaluated through several key parameters. The table below summarizes comparative data between bisulfite and enzymatic conversion approaches:
Table 1: Performance Comparison of DNA Conversion Methods [48]
| Parameter | Bisulfite Conversion (BC) | Enzymatic Conversion (EC) |
|---|---|---|
| Conversion Efficiency | Limit of reproducible conversion: 5 ng | Limit of reproducible conversion: 10 ng |
| DNA Recovery | Structurally overestimated (2.3 ± 0.7) | Lower recovery (0.7 ± 0.2) |
| DNA Fragmentation | High with degraded DNA (14.4 ± 1.2) | Low-medium with degraded DNA (3.3 ± 0.4) |
| DNA Input Range | 0.5-2000 ng | 10-200 ng |
| Protocol Time | 12-16 hours (including incubation) | 6 hours total |
| Cost per Conversion | €2.91 | €6.41 |
Table 2: Quality Control Metrics for MethylationEPIC BeadChip [49]
| QC Metric | Below Threshold | Pass Threshold | High Quality | Recommended Mitigation |
|---|---|---|---|---|
| Percentage of Failed Probes | >10% | 1-10% | ≤1% | Ensure optimal DNA input for bisulfite conversion; optimize PCR conditions |
| Beta Value Distribution | >2 peaks | 2 peaks | 2 clear peaks | Remove unreliable probes; eliminate background contamination |
| Percent of CpG Methylation | <20% or >80% | 20-80% | Within expected biological range | Repeat bisulfite conversion and whole genome amplification |
Troubleshooting Guides & FAQs
Conversion Efficiency Issues
Question: How can I troubleshoot low bisulfite conversion efficiency in my sperm DNA samples?
Answer: Low conversion efficiency leads to false positive methylation calls and can result from several factors [50]:
- Incomplete conversion: Ensure bisulfite treatment uses optimal temperature, incubation time, and salt concentrations. Extended incubation may be needed for highly compacted sperm DNA.
- DNA purity: Verify that your DNA sample is free of EDTA, which chelates metals and inhibits the conversion reaction. Use nuclease-free water or recommended elution buffers [51].
- Protocol optimization: For the EZ DNA Methylation kit (Zymo Research), ensure the 16-hour incubation is performed at the correct temperature and protected from light.
Question: What are the specific troubleshooting steps for low oxidation efficiency in enzymatic conversion?
Answer: For NEBNext Enzymatic Methyl-seq kits, low oxidation efficiency (pUC19 CpG methylation below 96%) can be addressed by [51]:
- Using fresh TET2 Reaction Buffer Supplement (resuspended less than 4 months)
- Ensuring accurate pipetting of viscous reagents and sufficient mixing
- Adding fresh DTT at the correct concentration
- Properly handling Fe(II) solution - pipette accurately using a P2 pipette tip and use within 15 minutes of dilution
- Avoiding addition of Fe(II) to the TET2 master mix - instead, mix sample with oxidation reagents before Fe(II) addition
Library Preparation Challenges
Question: Why is my library yield low after bisulfite conversion and how can I improve it?
Answer: Low library yields result from DNA degradation during conversion and suboptimal cleanup:
- Minimize degradation: For BC, consider reduced incubation time or switching to EC for fragile samples [48].
- Optimize bead cleanup: Avoid overdrying beads, ensure complete ethanol removal, and optimize elution conditions [51].
- Input DNA quality: Use high-quality sperm DNA with minimal somatic contamination. For sperm samples, implement somatic cell lysis buffer treatment and microscopic examination to eliminate contaminating cells [30].
Question: How can I prevent high fragmentation in bisulfite-converted sperm DNA?
Answer: DNA fragmentation is inherent to bisulfite chemistry but can be managed:
- Use enzymatic conversion: For precious sperm samples, EC causes significantly less fragmentation (3.3 ± 0.4) compared to BC (14.4 ± 1.2) [48].
- Optimize input DNA: Use the higher end of the recommended input range when possible.
- Adjust amplicon size: Design PCR amplicons ≤200bp for bisulfite-converted DNA, as larger amplicons may not amplify efficiently due to strand breaks [10].
Sperm-Specific Considerations
Question: What specialized steps are needed for sperm epigenetic studies to ensure accurate results?
Answer: Sperm samples present unique challenges requiring specific quality controls:
- Somatic cell contamination: Implement a comprehensive contamination control plan including [30]:
- Microscopic examination pre- and post-processing
- Somatic cell lysis buffer (SCLB: 0.1% SDS, 0.5% Triton X-100) treatment
- Application of a 15% cutoff during data analysis using specific CpG biomarkers
- Biomarker verification: Utilize identified 9,564 CpG sites that show high methylation in blood (>80%) but low methylation in sperm (<20%) to detect residual somatic contamination [30].
- DNA extraction optimization: Sperm chromatin compaction requires specialized lysis protocols for complete DNA recovery.
Experimental Protocols & Workflows
Assessing Conversion Efficiency
Protocol: qBiCo Multiplex qPCR Assay for Conversion Performance [48]
This method evaluates conversion efficiency, converted DNA recovery, and fragmentation:
Assay Design:
- Genome-wide conversion efficiency: Two assays targeting genomic and converted versions of the multi-copy human L1 repetitive element (LINE-1)
- Converted DNA concentration: One assay targeting the converted version of the single-copy hTERT gene
- Converted DNA fragmentation: Additional assays assessing DNA integrity
Procedure:
- Perform multiplex qPCR on converted DNA samples
- Calculate conversion efficiency by comparing genomic vs. converted L1 signals
- Determine recovery by quantifying converted hTERT relative to input DNA
- Assess fragmentation through size-dependent amplification patterns
Quality Thresholds:
- Conversion efficiency: >99% for most applications
- Limit of reproducible conversion: 5ng for BC, 10ng for EC
- Recovery index: 0.7-2.3, depending on method
Protocol: Computational Assessment of Bisulfite Conversion Ratio [52]
The BCREval method uses telomeric repeats as internal controls:
Principle: Telomeric TTAGGG repeats contain non-CpG cytosines that should be fully unmethylated, serving as native spike-in controls.
Workflow:
- Extract telomeric reads from WGBS data (minimal 8 repeat blocks)
- Calculate unconversion ratio (UCR) at non-CpG sites
- Determine bisulfite conversion ratio: BCR = 1 - UCR
Implementation:
- Requires only forward FASTQ files from WGBS
- Much faster than alignment-based methods (30x faster than Bismark)
- Effective for quality control in high-throughput workflows
DNA Conversion and QC Workflow
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Bisulfite Conversion Quality Control
| Reagent/Kit | Function | Application Notes |
|---|---|---|
| EZ DNA Methylation Kit (Zymo Research) | Bisulfite conversion | Popular for Illumina Infinium arrays; requires 16h incubation [48] |
| NEBNext Enzymatic Methyl-seq Kit | Enzymatic conversion | Gentler alternative to BC; uses TET2 oxidation and APOBEC deamination [48] |
| qBiCo Assay Components | Conversion performance assessment | Multiplex qPCR for efficiency, recovery, fragmentation indexes [48] |
| Somatic Cell Lysis Buffer | Sperm purification | Removes contaminating somatic cells (0.1% SDS, 0.5% Triton X-100) [30] |
| Lambda DNA Controls | Conversion efficiency monitoring | Unmethylated control for assessing conversion efficiency [53] |
| pUC19 Methylated Control | Oxidation efficiency verification | For enzymatic conversion efficiency assessment [51] |
| Aminoacyl tRNA synthetase-IN-1 | Aminoacyl tRNA synthetase-IN-1, MF:C16H25N7O7S, MW:459.5 g/mol | Chemical Reagent |
| Leu-AMS | Leu-AMS|Leucyl-tRNA Synthetase Inhibitor|mTORC1 Research |
Standardization of quality control benchmarks for bisulfite conversion efficiency and library preparation is fundamental for advancing sperm epigenetic research across laboratories. The integration of robust quantitative metrics, sperm-specific contamination controls, and both experimental and computational verification methods provides a comprehensive framework for reliable DNA methylation analysis. As enzymatic conversion technologies continue to evolve, they offer promising alternatives to traditional bisulfite treatment, particularly for compromised sperm samples. Implementation of these troubleshooting guides and quality control protocols will enhance reproducibility and data quality in male fertility studies, ultimately supporting more accurate diagnostic and therapeutic development in andrology research.
Integrating multi-omic data with functional sperm parameters presents significant computational and experimental challenges for researchers working on male fertility. The inherent heterogeneity of data originating from different biological layers—genome, epigenome, transcriptome, and proteome—creates substantial bottlenecks in analysis and interpretation [54]. Furthermore, technical variations in sample collection, processing, and analysis across laboratories complicate the standardization necessary for robust, reproducible research.
This technical support center addresses these challenges by providing practical troubleshooting guides and frequently asked questions framed within the context of standardizing sperm epigenetic protocols. Our guidance draws from recent advances in the field, including studies that have successfully identified biomarkers for bull fertility through multi-omics integration [55] and research examining how environmental factors influence the sperm epigenome [26].
Frequently Asked Questions (FAQs)
FAQ 1: What are the most critical pre-analytical factors that affect sperm epigenomic data? Multiple pre-analytical factors significantly impact data quality. Sperm storage conditions—particularly prolonged in vitro storage—adversely affect DNA methylation patterns, reduce sperm motility, and increase DNA fragmentation [56]. Lifestyle and environmental exposures, including paternal obesity, smoking, stress, and endocrine-disrupting chemicals, alter sperm DNA methylation, histone retention patterns, and small non-coding RNA profiles [26]. Additionally, incomplete somatic cell removal during sample preparation contaminates epigenomic profiles since somatic cells have distinct epigenetic signatures compared to sperm cells [57].
FAQ 2: Which functional sperm parameters show the strongest correlation with epigenetic marks? Research indicates that sperm motility and velocity parameters (VCL and VAP) demonstrate significant correlation with epigenetic alterations, particularly DNA methylation changes [56]. DNA integrity shows strong associations with aberrant methylation at imprinted genes such as H19, MEST, and SNRPN [4]. Sperm concentration correlates with methylation levels of genes involved in spermatogenesis, including DAZL and SOX30 [4]. Furthermore, fertilization capacity links to global methylation patterns and specific small RNA profiles, with hypermethylation in promoter regions of genes like PLAG1, PAX8, and DIRAS3 negatively impacting motility and morphology [4].
FAQ 3: What are the common pitfalls when integrating different omics datasets? Researchers frequently encounter several integration pitfalls: failing to properly standardize and harmonize data from different technological platforms and measurement units [58]; designing integration pipelines from a data curator's perspective rather than the end-user's needs, reducing utility [58]; insufficient statistical power from small sample sizes relative to the high dimensionality of multi-omics data [54]; and misinterpreting correlations as causal relationships without functional validation [54] [59].
FAQ 4: How can we address the challenge of transgenerational epigenetic inheritance studies? Key challenges in this area include distinguishing true transgenerational inheritance (through multiple generations) from intergenerational effects (direct exposure of germ cells) [59]; controlling for genetic predisposition and maternal environmental confounders [26]; and understanding molecular mechanisms that maintain epigenetic marks through global reprogramming events after fertilization [26]. Technical approaches should implement controlled, multi-generational animal models, utilize multi-omics integration to identify consistent signals across biological layers, and develop standardized protocols for germ cell collection and analysis across collaborating laboratories [59].
Troubleshooting Guides
Poor Correlation Between DNA Methylation and Transcriptomic Data
Problem: After integrating sperm DNA methylation and transcriptomic data, expected inverse correlations between promoter methylation and gene expression are not observed.
Solutions:
- Verify data quality and preprocessing: Ensure bisulfite conversion rates exceed 99.45% and RNA integrity numbers (RIN) are suitable for sperm RNA (typically >7) [56]. Standardize normalization methods across datasets to remove technical artifacts [58].
- Expand genomic context analysis: Examine methylation in gene bodies and enhancer regions, not just promoters, as gene body methylation can have different relationships with expression [56]. Incorporate histone retention maps since retained nucleosomes in sperm are enriched at developmental regulators and may confimple methylation-expression relationships [57].
- Account for biological complexity: Consider time-lagged effects where epigenetic changes may affect expression at different developmental stages [56]. Include additional omics layers like proteomics to verify functional outcomes, as mRNA levels may not directly correlate with protein abundance [54].
Technical Variance Obscuring Biological Signals in Multi-Omic Data
Problem: Batch effects and technical variance dominate the integrated dataset, masking biologically relevant signals.
Solutions:
- Implement batch correction algorithms: Apply ComBat or other batch effect correction tools specifically designed for omics data [58]. Include experimental batches as covariates in differential analysis models.
- Standardize protocols across participating labs: Establish standardized SOPs for sample collection, storage duration, and processing [56]. Use common reference materials across batches and laboratories to enable normalization.
- Design studies with integration in mind: Ensure balanced sample allocation across processing batches [58]. Collect comprehensive metadata including processing dates, reagent lots, and operator information to facilitate technical variance modeling.
Inter-Laboratory Variation in Sperm Epigenetic Measurements
Problem: Significant differences emerge when the same sample is analyzed across different laboratories, hindering protocol standardization.
Solutions:
- Establish reference standards: Develop and distribute reference sperm samples with characterized epigenetic profiles for inter-laboratory calibration [58]. Implement internal controls spiked into each sample, such as synthetic methylated DNA standards.
- Harmonize data processing pipelines: Create containerized analysis pipelines (Docker/Singularity) to ensure consistent bioinformatic processing [58]. Adopt common quality control thresholds and reporting standards.
- Cross-validate findings: Confirm key results using multiple technologies (e.g., WGBS and EPIC array for methylation) [4]. Organize ring trials where laboratories exchange samples and compare results.
Table 1: Sperm Functional Parameters and Their Associated Epigenetic Alterations
| Functional Parameter | Measurement Method | Associated Epigenetic Changes | Impact on Function |
|---|---|---|---|
| Motility | Computer-assisted sperm analysis (CASA) | Hypermethylation of PLAG1, PAX8, DIRAS3 promoters [4] | Reduced progressive motility |
| DNA Integrity | TUNEL assay | Hypomethylation of H19 imprinting control region [4] | Increased DNA fragmentation index |
| Concentration | Hemocytometer/ CASA | Aberrant methylation of DAZL, SOX30 [4] | Impaired spermatogenesis |
| Morphology | Kruger strict criteria | Hypermethylation of MEST, HRAS [4] | Increased teratozoospermia |
| Fertilization Capacity | In vitro fertilization assays | Global methylation changes; sncRNA profile alterations [26] | Reduced embryo development rates |
Table 2: Multi-Omics Integration Methodologies and Their Applications
| Integration Method | Data Types Combined | Key Applications | Software/Tools |
|---|---|---|---|
| Multiple Factor Analysis | Genotypes, DNA methylation, sncRNAs, semen parameters [55] | Biomarker identification for bull fertility | R: FactoMineR |
| Machine Learning (Lasso, Random Forest) | SNPs, CpG methylation, miRNAs [55] | Feature selection for fertility prediction | Python: scikit-learn |
| Multi-layer Networks | Genome, epigenome, transcriptome, proteome [54] | Modeling biological mechanisms | Cytoscape, OmicsNet |
| Comparative Pathway Analysis | DNA methylome, transcriptomic, proteomic [56] | Identifying storage-induced alterations | GSEA, clusterProfiler |
| Style Transfer Methods | Heterogeneous omics datasets [58] | Data harmonization across platforms | Conditional variational autoencoders |
Experimental Workflows and Signaling Pathways
Multi-Omics Integration Workflow for Sperm Analysis
Environmental Impact on Sperm Epigenome
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Sperm Multi-Omics Studies
| Reagent/Category | Specific Examples | Function & Application |
|---|---|---|
| Bisulfite Conversion Kits | EZ DNA Methylation kits | Convert unmethylated cytosines to uracils for methylation analysis [56] |
| Protamine Removal Agents | Dithiothreitol (DTT), Triton X-100 | Decondense sperm chromatin for DNA and protein extraction [57] |
| Sperm Storage Media | Artificial seminal plasma | Maintain sperm viability during short-term storage studies [56] |
| DNA Methyltransferases Inhibitors | 5-aza-2'-deoxycytidine | Experimental modulation of methylation patterns [4] |
| Histone Modification Antibodies | H4K5ac, H3K4me2/3 | Mapping histone retention sites in sperm chromatin [57] [26] |
| sncRNA Isolation Kits | miRNeasy, mirVana | Enrichment of small non-coding RNAs from sperm [55] |
| Somatic Cell Lysis Buffers | SDS-based lysis solutions | Remove somatic cell contamination prior to epigenomic analysis [57] |
| Viability Stains | Hoechst 33342, Propidium Iodide | Assess membrane integrity and sperm quality [56] |
| Zofenopril | Zofenopril, CAS:81872-10-8, MF:C22H23NO4S2, MW:429.6 g/mol | Chemical Reagent |
| EP1013 | EP1013, MF:C18H23FN2O6, MW:382.4 g/mol | Chemical Reagent |
Navigating Technical Challenges and Ensuring Assay Reproducibility
Frequently Asked Questions (FAQs)
1. Why is controlling pre-analytical variation so critical in sperm epigenetics research? Pre-analytical errors account for up to 75% of all laboratory errors, and variations in donor selection or specimen collection can significantly alter the epigenetic profile of a sample, compromising data reliability and reproducibility across studies [60] [61].
2. What is the recommended ejaculatory abstinence (EA) period for sperm epigenetic studies? While traditional WHO guidelines for semen analysis recommend 2-7 days, recent evidence suggests that a shorter abstinence period of approximately 24 hours (1 day) is associated with better sperm quality in terms of motility, acrosome integrity, mitochondrial activity, and, crucially, nuclear DNA integrity [62]. Samples collected after 1 day of EA showed a decrease in oxidative activity compared to those after 4 days, which helps preserve DNA quality [62].
3. How does donor selection influence research outcomes? The epigenetic age of sperm, determined by DNA methylation patterns, is highly donor-specific [63]. Furthermore, sperm from men seeking infertility treatment can show significantly different epigenetic variability compared to fertile sperm donors, which directly impacts reproductive potential and research results [64]. Therefore, rigorous and standardized donor screening criteria are essential.
4. What are the key donor factors to control for during selection? Key factors include age, health status (excluding systemic diseases, recent fever, etc.), lifestyle habits (smoking, alcohol consumption), and medication use [62] [65]. These factors are known to influence epigenetic markers and should be carefully documented [66].
Troubleshooting Guide: Common Pre-Analytical Challenges
Problem: Inconsistent DNA Methylation Results After Ejaculation
Potential Cause: Inappropriate ejaculatory abstinence period leading to oxidative stress and DNA fragmentation [62]. Solution: Standardize the EA period to 24 hours for research focused on DNA quality and epigenetics. Ensure participants are thoroughly instructed and reminded to adhere to the abstinence period before sample collection [62].
Problem: High Inter-Donor Variability in Epigenetic Profiles
Potential Cause: Inadequate donor screening and selection criteria [63] [64]. Solution: Implement strict, documented inclusion and exclusion criteria for donors. The table below summarizes key criteria based on research evidence [62]:
Table: Essential Donor Inclusion and Exclusion Criteria
| Category | Inclusion Criteria | Exclusion Criteria |
|---|---|---|
| Health Status | Healthy | History of systemic diseases (e.g., cancer, diabetes), urogenital surgeries, fever in the 90 days before collection [62] |
| Lifestyle | History of smoking, excessive alcohol or drug consumption [62] | |
| Semen Parameters | Azoospermia, sperm concentration < 5 × 10^6/mL, seminal volume < 1 mL [62] | |
| Other | Age between 20-45 years [62] | Body mass index (BMI) > 35 kg/m² [62] |
Problem: Degraded Sample Quality Upon Arrival at the Lab
Potential Cause: Improper sample transport conditions [65]. Solution: Transport semen samples to the laboratory within one hour of ejaculation. Keep the specimen container upright in a plastic bag, with the lid securely tightened, and maintain it as close to body temperature as possible. Do not place it in pockets or bags, as temperature fluctuations inactivate sperm [65].
Experimental Protocols for Standardization
Protocol 1: Standardized Procedure for Semen Sample Collection
This protocol is designed to minimize pre-analytical variation for epigenetic studies [62] [65].
Participant Preparation and Instruction:
- Provide donors with detailed, written instructions.
- Instruct donors to maintain 24 hours (± 2 hours) of ejaculatory abstinence.
- Direct donors to limit caffeine, tobacco, and alcohol for the 10 days preceding collection.
- Confirm the absence of fever or hot tub use in the recent past.
- One week before collection, confirm the schedule and abstinence requirements via a phone call or email [62].
Sample Collection:
- Collect the sample on-site in a private room to ensure proper identity verification and minimize transport time.
- Provide a sterile, wide-mouthed container. Do not use regular condoms or lubricants not provided by the lab.
- The donor should wash hands and penis thoroughly with soap and water before collection.
- Ejaculate directly into the container, ensuring the first portion of the ejaculate (sperm-rich) is captured.
Sample Handling and Transport:
- Securely fasten the lid immediately after collection. Label the container with the donor's name, date of birth, and exact time of collection.
- Transport the sample to the laboratory at room temperature, ensuring analysis begins within one hour of ejaculation [65].
Protocol 2: Targeted DNA Methylation Analysis via Bisulfite Sequencing
This methodology, adapted from forensic epigenetic age prediction studies, allows for precise measurement of age-correlated CpG sites in sperm DNA [63].
- DNA Extraction: Extract genomic DNA from semen samples using a standardized phenol-chloroform or column-based method. Quantify DNA concentration and assess purity.
- Bisulfite Conversion: Treat 500 ng of genomic DNA with sodium bisulfite using a commercial kit (e.g., EZ DNA Methylation-Lightning Kit). This process converts unmethylated cytosines to uracils, while methylated cytosines remain unchanged.
- Targeted Amplification: Design PCR primers to amplify specific age-correlated genomic regions (e.g., CpGs in genes like SH2B2, EXOC3, IFITM2, GALR2, and FOLH1B). Use a hot-start polymerase for high specificity [63].
- Library Preparation and Sequencing: Prepare sequencing libraries from the amplified products using a commercial kit for massively parallel sequencing (MPS). Perform paired-end sequencing on an appropriate MPS platform.
- Data Analysis:
- Align sequencing reads to a bisulfite-converted reference genome.
- Calculate methylation levels at each targeted CpG site as the percentage of reads showing a cytosine compared to the total reads at that position.
- Input the methylation percentages into a validated prediction model (e.g., a linear regression model) to estimate epigenetic age or other parameters of interest [63].
Data Presentation: Impact of Pre-Analytical Variables
Table: Quantitative Impact of Ejaculatory Abstinence Period on Semen Parameters
| Parameter | 1-Day Abstinence | 4-Day Abstinence | Biological Implication for Epigenetics |
|---|---|---|---|
| Semen Volume | Decreased [62] | Increased [62] | Less relevant for DNA methylation analysis. |
| Sperm Total Number | Decreased [62] | Increased [62] | Affects available DNA yield. |
| Sperm Motility | Better [62] | Reduced [62] | Correlates with overall sperm health. |
| Nuclear DNA Integrity | Higher [62] | Lower [62] | Critical: Directly impacts quality for epigenetic assays. |
| Intracellular Oxidative Activity | Lower [62] | Higher [62] | Critical: Oxidative stress causes DNA damage and can alter methylation. |
Workflow Visualization
Diagram Title: Pre-Analytical Workflow for Sperm Epigenetics
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Materials for Sperm Epigenetic Analysis
| Item | Function/Benefit | Example/Note |
|---|---|---|
| Bisulfite Conversion Kit | Chemically converts unmethylated cytosine to uracil for downstream methylation detection. | Essential for methods like bisulfite sequencing. Use kits validated for sperm DNA. |
| Targeted Bisulfite MPS Assay | Enables highly sensitive, simultaneous analysis of multiple age-correlated CpG sites from low-quality/quantity DNA. | Overcomes limitations of microarrays for forensic-type samples [63]. |
| Protamine Displacement Assay | Evaluates the efficiency of histone-to-protamine replacement, a key epigenetic event in spermatogenesis. | Assesses sperm chromatin maturity, linked to DNA integrity [66]. |
| Sterile Semen Collection Cup | Provides a non-toxic, sterile container for sample collection to avoid contamination and spermicide exposure. | Do not use regular condoms. Wide-mouthed cups are preferred [65]. |
| Sperm DNA Integrity Kit | Quantifies the level of DNA fragmentation in sperm, a key quality marker. | e.g., SCD (Sperm Chromatin Dispersion) or TUNEL assay kits. |
| Validated CpG Marker Panel | A minimal set of highly age-predictive DNA methylation markers for robust modeling. | e.g., Panel including CpGs from SH2B2, EXOC3, IFITM2, GALR2, FOLH1B [63]. |
| p38 MAPK-IN-2 | p38 MAPK-IN-2|p38 Inhibitor|For Research Use | p38 MAPK-IN-2 is a potent p38 MAPK inhibitor for cell signaling research. This product is For Research Use Only and not intended for diagnostic or personal use. |
| Jak-IN-10 | Jak-IN-10, MF:C20H18FN5O3S, MW:427.5 g/mol | Chemical Reagent |
Mitigating Contamination from Somatic Cells and Immature Germ Cells
In sperm epigenetic research, the accuracy of data interpretation is paramount. A significant threat to this accuracy is contamination from somatic cells (such as leukocytes) and immature germ cells, which possess vastly different epigenetic landscapes. Sperm DNA is characteristically hypomethylated in many promoter regions, while somatic cell DNA is typically hypermethylated in these same areas. Consequently, even low-level contamination can introduce a false hypermethylation signal, leading to erroneous conclusions about sperm quality, fertility status, and the potential for transgenerational inheritance [67]. This guide provides standardized, actionable protocols to identify, quantify, and eliminate this contamination, ensuring the integrity of your sperm epigenetic data.
Frequently Asked Questions (FAQs) and Troubleshooting Guides
Q1: Why is somatic cell contamination a particular problem for oligozoospermic samples? Semen samples from oligozoospermic (low sperm count) individuals are inherently more vulnerable to significant somatic cell contamination. As the sperm count decreases, the relative proportion and impact of contaminating somatic cells increase dramatically. A small, fixed number of somatic cells will constitute a much higher percentage of the total DNA in a sample with few sperm, thereby exerting a greater influence on the final epigenetic measurements and increasing the risk of a misleading proxy methylation signal [67].
Q2: After somatic cell lysis buffer (SCLB) treatment, microscopic examination shows no somatic cells. Is my sample now completely clean? Not necessarily. While microscopic examination is a crucial quality control step, it has a detection limit. It is challenging to reliably detect somatic cell contamination when it falls below approximately 5% of the sperm number [67]. Therefore, a sample can appear clean under the microscope yet still contain enough somatic cells to bias sensitive downstream analyses like DNA methylation sequencing or array analysis. A multi-faceted approach is required for guaranteed purity.
Q3: What is the single most effective step I can add to my protocol to control for hidden contamination? Incorporate a bioinformatic checkpoint using known somatic cell-specific CpG markers. By analyzing the methylation levels at a panel of genomic loci that are highly methylated in somatic cells but hypomethylated in sperm, you can detect and quantify contamination in silico after data generation. Applying a 15% cut-off during data analysis to exclude samples with contamination signals above this threshold is an effective final safeguard [67].
Q4: My research involves other techniques like flow cytometry. Are there integrated methods for quality assessment? Yes, flow cytometry is a powerful tool for immunophenotyping and can be adapted for cell population quantification. While the provided search results focus on its use in immunology [68] [69], the principles are transferable. A robust strategy involves using a paired assessment approach where a sample is split for different analyses (e.g., one portion for epigenetic work and another for flow cytometry with specific cell surface markers) to cross-validate cell population purity [69]. Always include proper controls, such as Fluorescence-Minus-One (FMO) controls, to ensure gating accuracy [68].
Comprehensive Experimental Protocols
Protocol 1: Somatic Cell Lysis and Microscopic Examination
This protocol outlines the initial physical removal of somatic cells from semen samples.
- Objective: To significantly reduce the somatic cell population in fresh semen samples through chemical lysis and verify the reduction via microscopy.
- Materials: Fresh semen sample, 1X Phosphate-Buffered Saline (PBS), Somatic Cell Lysis Buffer (SCLB: 0.1% SDS, 0.5% Triton X-100 in ddH2O), centrifuge, microscope.
- Procedure:
- Wash: Wash the fresh semen sample twice with 1X PBS by centrifugation at 200 g for 15 minutes at 4°C.
- Initial Inspection: Resuspend the pellet and inspect an aliquot under a microscope (e.g., 20X objective). Record the presence and approximate number of somatic cells and count the sperm.
- Lysis: Incubate the sample with freshly prepared SCLB for 30 minutes at 4°C.
- Post-Lysis Inspection: Centrifuge the sample to obtain a pellet, resuspend in PBS, and check again under the microscope for the presence of somatic cells.
- Repeat if Necessary: If any somatic cells are detected, repeat the SCLB treatment step.
- Final Wash: Once no somatic cells are visible, pellet the sperm by centrifugation and perform a final wash with PBS to obtain a highly pure sperm population [67].
Protocol 2: Validating Purity via Somatic DNA Methylation Biomarkers
This protocol provides a method for validating sample purity after wet-lab processing using bioinformatic analysis.
- Objective: To detect and quantify residual somatic DNA contamination by analyzing specific CpG methylation biomarkers.
- Materials: DNA extracted from the purified sperm sample, dataset from a DNA methylation array (e.g., Infinium Human Methylation 450K BeadChip) or sequencing.
- Procedure:
- Data Generation: Process your sperm DNA through your standard genome-wide methylation analysis platform (e.g., microarray or bisulfite sequencing).
- Biomarker Analysis: In your resulting data, extract the methylation beta-values for a predefined set of somatic-specific CpG markers. Research has identified 9,564 CpG sites that are highly methylated in blood (>80%) and minimally methylated in sperm (<20%) and are not linked to infertility, making them ideal contamination markers [67].
- Contamination Assessment: Calculate the average methylation level across these biomarker CpGs for each sample.
- Quality Threshold: Apply a strict cut-off. Samples showing an average methylation level of >15% at these somatic-specific markers should be considered contaminated and excluded from final analysis, as the signal likely represents proxy methylation from somatic DNA rather than true sperm DNA methylation [67].
Data Presentation: Key Biomarkers and Quality Thresholds
Table 1: Selected Somatic Cell-Specific CpG Methylation Biomarkers for Contamination Assessment. This table lists a subset of potential markers from the larger panel of 9,564. High methylation at these loci in a sperm sample indicates somatic cell contamination [67].
| CpG Probe ID | Genomic Location | Gene Association | Methylation in Blood | Methylation in Sperm |
|---|---|---|---|---|
| Example 1 | Chromosome 1: 1,234,567 | EXAMPLE1 | >80% | <20% |
| Example 2 | Chromosome 5: 67,890,123 | EXAMPLE2 | >80% | <20% |
| Example 3 | Chromosome 12: 34,567,890 | EXAMPLE3 | >80% | <20% |
Table 2: Decision Matrix for Sample Inclusion Based on Contamination Checks. This workflow ensures only high-quality, uncontaminated data is used for final analysis.
| Checkpoint | Method | Acceptance Criterion | Action if Failed |
|---|---|---|---|
| Initial Quality Check | Microscopic Examination | No somatic cells visible. | Repeat SCLB treatment. |
| Final Quality Gate | Somatic CpG Biomarker Analysis | Average β-value < 0.15 (15%) | Exclude sample from dataset. |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Sperm Contamination Mitigation.
| Reagent / Material | Function / Application | Example / Specification |
|---|---|---|
| Somatic Cell Lysis Buffer (SCLB) | Selectively lyses somatic cells (e.g., leukocytes) while leaving sperm cells intact for subsequent analysis. | 0.1% SDS, 0.5% Triton X-100 in ddHâ‚‚O [67]. |
| CpG Methylation Biomarker Panel | A set of genomic coordinates used for bioinformatic detection of somatic DNA contamination in sperm methylation data. | Panel of 9,564 CpG sites hypermethylated in blood vs. sperm [67]. |
| Flow Cytometry Antibody Panel | Antibodies for surface markers to identify and quantify specific immune cell populations in a heterogeneous sample. | e.g., CD45-APC (leukocytes), CD3-PE (T cells) [69]. |
| Fluorescence-Minus-One (FMO) Controls | Critical controls for flow cytometry that help establish correct positive/negative gates for each fluorescent channel, ensuring accurate immunophenotyping. | Sample stained with all antibodies except one [68]. |
Workflow and Pathway Visualizations
The following diagram illustrates the comprehensive, multi-step workflow for ensuring sperm sample purity, from physical processing to computational validation.
This workflow provides a logical, step-by-step guide for the contamination mitigation process. The following diagram details the specific signaling consequence of contamination, showing how somatic cell DNA leads to incorrect scientific conclusions.
Optimizing Bioinformatics Pipelines for Consistent DMR and sncRNA Identification
Troubleshooting Guides
Low-Quality sncRNA Identification
Problem: Your pipeline fails to identify a diverse range of small noncoding RNAs (sncRNAs), showing strong bias toward microRNAs while missing other sncRNA types.
Explanation: Standard small RNA-seq methods have inherent limitations because many sncRNAs contain unique chemical modifications at their ends that can interfere with adapter ligation and block reverse transcription, preventing their detection [70]. This technical bias means only a subset of small RNAs (primarily miRNAs) are reliably detected, while others remain hidden.
Solution: Implement specialized enzymatic pre-treatment and updated analysis pipelines.
- Wet-Lab Protocol: Use PANDORA-seq, which employs a two-step enzymatic treatment prior to sequencing [70].
- T4 Polynucleotide Kinase (T4 PNK): Modifies RNA molecules by adding or removing phosphate groups at their ends, making them more suitable for adapter ligation.
- AlkB (a bacterial demethylase): Removes RNA modifications that would otherwise block reverse transcription, allowing for more efficient cDNA synthesis [70].
- Bioinformatic Protocol: Utilize the SPORTS 1.1 pipeline or SCRAP pipeline for data analysis. SCRAP is specifically optimized for the distinctive characteristics of chimeric sncRNA sequencing reads and consists of two parts [71]:
- Read processing and alignment: Specifically optimized for chimeric small RNA sequencing data.
- Peak calling and annotation: Designed to broaden accessibility for processing small chimeric RNA sequencing data [71].
Inconsistent Differentially Methylated Region (DMR) Calls
Problem: DMR identification yields different results when using different analysis tools or workflows, leading to irreproducible biomarker discovery.
Explanation: The choice of analysis pipeline for DNA methylation-based marker discovery is crucial and varies across different biological contexts [72]. Numerous computational tools and parameter options exist for identifying DMRs from array or sequencing data, and there is no universal "best" workflow. Performance depends on dataset characteristics, such as tissue type or the specific methylation patterns being studied [72].
Solution: Systematically benchmark and select analysis workflows using standardized simulated data.
- Benchmarking Methodology: Use a simulation method like TASA (Tissue Aware Simulation Approach) that uses reference methylation data to simulate known DMRs while accounting for biological and technical noise [72].
- Workflow Selection: Comprehensively assess different data analysis workflows using both real and simulated data to determine the optimal combination of tools for your specific context (e.g., specific tissue type or disease state) [72]. The TASA method can simulate 12 different contexts to suggest suitable workflows [72].
Pipeline Execution Failures
Problem: The bioinformatics pipeline fails to run successfully, halting with errors related to tool compatibility, data input, or system resources.
Explanation: Bioinformatics pipelines are complex workflows that integrate various tools and algorithms. Errors can arise from multiple sources, including data quality issues, tool compatibility conflicts, computational bottlenecks, or insufficient documentation [73].
Solution: Follow a structured troubleshooting approach to isolate and resolve the issue.
- Identify the Problem: Check error logs and pipeline outputs to pinpoint the specific error message [74].
- Isolate the Stage: Determine which pipeline component is causing the problem [73]. Common failure points include:
- Resolve the Issue:
- For data quality issues, use tools like FastQC and Trimmomatic to identify and remove contaminants [73].
- For tool compatibility, ensure all required tools and their specific versions are installed. Using containerization (e.g., Docker, Singularity) can mitigate this [74].
- For computational bottlenecks, consider migrating to a cloud platform with scalable computing power or optimizing your pipeline's resource allocation [73].
Frequently Asked Questions (FAQs)
What is the primary purpose of bioinformatics pipeline troubleshooting? The primary purpose is to identify and resolve errors or inefficiencies in computational workflows, ensuring the accuracy, reliability, and reproducibility of biological data analysis. Effective troubleshooting prevents the introduction of biases, reduces processing time and costs, and ensures results can be replicated by other researchers [73].
How can I start building a robust bioinformatics pipeline for sperm epigenetics? Begin by clearly defining your research objectives and the type of data to be analyzed. Select tools and algorithms tailored to your specific dataset and goals. Design the workflow by mapping out all pipeline stages, including data input, processing, analysis, and output. Test the pipeline on small-scale datasets to identify potential issues before running full analyses [73].
What are the most common tools used in bioinformatics pipeline troubleshooting? Commonly used tools include:
- Workflow Management: Nextflow, Snakemake, Galaxy [73]
- Data Quality Control: FastQC, MultiQC, Trimmomatic [73]
- Version Control: Git [73]
- sncRNA Analysis: SCRAP, SPORTS [71] [70]
- Bisulfite Sequencing Alignment: Bismark, BSMAP [75]
- Differential Methylation Analysis: Minfi, ChAMP, RnBeads [72]
How do I ensure the accuracy and reproducibility of my DMR/sncRNA analysis? Validate your results with known datasets or alternative methods. Cross-check outputs using different tools or parameters. Maintain detailed documentation of every change made to the pipeline, including software versions, parameters, and configurations. Use version control systems like Git to track changes and ensure reproducibility [73].
What industries benefit the most from optimized bioinformatics pipelines? Healthcare and medicine (e.g., genomic medicine, drug discovery, cancer research), environmental studies (e.g., monitoring biodiversity, tracking pathogens), agriculture, and biotechnology are among the industries that rely heavily on robust bioinformatics pipelines [73].
Experimental Protocols & Workflows
Detailed Protocol: sncRNA Identification via PANDORA-seq
Principle: Overcome sequencing biases introduced by RNA modifications to achieve a more comprehensive snapshot of the small RNA transcriptome [70].
Procedure:
- RNA Preparation: Extract total RNA, including small RNA fractions, from sperm samples.
- Enzymatic Treatment:
- Treat RNA with T4 Polynucleotide Kinase (T4 PNK) to modify the 5' and 3' ends.
- Subsequently treat with AlkB to remove methylation modifications (e.g., m1A, m3C) that block reverse transcription [70].
- Library Construction: Construct sequencing libraries from the treated RNA using standard small RNA-seq library preparation protocols.
- Sequencing: Perform deep sequencing on an appropriate platform (e.g., Illumina).
- Bioinformatic Analysis: Analyze the resulting data using the SPORTS pipeline or the SCRAP pipeline to identify and characterize sncRNAs, including those derived from tRNA, rRNA, and other parental RNAs [70] [71].
Detailed Protocol: DMR Identification from Bisulfite Sequencing Data
Principle: Identify genomic regions that show statistically significant differences in methylation levels between experimental groups (e.g., control vs. treatment) using bisulfite-converted sequencing data [75].
Procedure:
- Data Preprocessing:
- Perform quality control on raw sequencing reads (FASTQ files) using FastQC or Trim Galore!.
- Trim low-quality bases and adapter sequences [75].
- Read Mapping:
- Align bisulfite-treated reads to a reference genome using a specialized aligner such as Bismark (a three-letter aligner) or BSMAP (a wildcard aligner) [75].
- Account for C-to-T conversions in the sequencing reads during alignment.
- Methylation Calling:
- Extract methylation information from aligned reads (BAM files).
- Calculate methylation levels (e.g., as a beta-value) for each cytosine in a CpG context by counting the ratio of C/(C+T) reads [75].
- Differential Methylation Analysis:
- Annotation and Interpretation:
- Annotate significant DMRs with genomic features (e.g., promoters, gene bodies, CpG islands).
- Integrate with other data types (e.g., gene expression) for functional interpretation.
Workflow Diagrams
sncRNA Analysis with SCRAP
DMR Identification Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 1: Essential reagents and tools for sncRNA and DMR analysis.
| Item | Function | Application |
|---|---|---|
| T4 PNK | Modifies RNA ends by adding/removing phosphate groups | Enables adapter ligation for modified sncRNAs in PANDORA-seq [70]. |
| AlkB | Bacterial demethylase that removes RNA modifications | Unblocks reverse transcription for a wider range of sncRNAs in PANDORA-seq [70]. |
| Sodium Bisulfite | Chemical that converts unmethylated cytosine to uracil | Distinguishes methylated from unmethylated cytosines in bisulfite sequencing [75]. |
| SCRAP Pipeline | Bioinformatic pipeline for analyzing chimeric sncRNA-seq data | Identifies miRNA-mRNA and other sncRNA-target interactions with high sensitivity [71]. |
| SPORTS Pipeline | Bioinformatic analysis tool | Used with PANDORA-seq data to identify and characterize a diverse set of sncRNAs [70]. |
| Bismark | Alignment tool for bisulfite sequencing reads | Accurately maps bisulfite-converted reads to a reference genome for methylation analysis [75]. |
| Minfi / ChAMP | R/Bioconductor packages | Comprehensive analysis of DNA methylation data, from preprocessing to DMR identification [72]. |
| TASA Simulator | Method for simulating methylome data with known DMRs | Benchmarks and selects optimal DMR analysis workflows for specific contexts [72]. |
Correcting for Batch Effects in Multi-Center Epigenetic Studies
Batch effects are systematic technical variations that are unrelated to the biological factors under investigation but can severely distort the results of your epigenetic studies. In the context of multi-center research aimed at standardizing sperm epigenetic protocols, these effects present a critical challenge that must be addressed to ensure data reliability and cross-laboratory reproducibility.
In epigenetic profiling, particularly DNA methylation analysis, batch effects can arise from differences in reagent lots, instrumentation, personnel, sequencing runs, sample preparation protocols, and bisulfite conversion efficiency [76] [77]. These technical variations can obscure true biological signals, leading to both false positives and false negatives in differential methylation analysis [78] [77]. For multi-center sperm epigenetic studies, where the goal is to identify consistent biomarkers across different laboratories, failing to correct for batch effects can compromise the entire standardization effort, potentially leading to irreproducible findings and incorrect biological conclusions [77].
The fundamental issue stems from the assumption that instrument readout intensity linearly represents analyte concentration. In practice, the relationship between actual methylation and measured values fluctuates across different experimental conditions, making measurements inherently inconsistent across batches [77]. Understanding, detecting, and correcting these artifacts is therefore essential for any successful multi-center epigenetic standardization initiative.
Troubleshooting Guide: Common Issues & Solutions
How do I detect batch effects in my DNA methylation data?
Problem: You suspect technical variations are affecting your methylation data but are unsure how to confirm their presence.
Solution: Implement both visual and quantitative assessment methods:
- Principal Component Analysis (PCA): Perform PCA on your raw methylation data (beta-values or M-values) and color points by batch. If samples cluster primarily by batch rather than biological group, significant batch effects are present [79]. This is the most common and intuitive detection method.
- t-SNE/UMAP Visualization: Create t-SNE or UMAP plots with cells labeled by batch. In the presence of uncorrected batch effects, cells from different batches will form separate clusters rather than mixing according to biological similarities [79].
- Quantitative Metrics: Calculate metrics such as the k-nearest neighbor batch effect test (kBET) or adjusted rand index (ARI). These provide objective measures of batch effect severity before and after correction [79].
Prevention Tip: Whenever possible, include technical replicates across batches and reference samples in your experimental design to facilitate batch effect detection and correction [78].
What should I do when my samples cluster by batch rather than biological group after analysis?
Problem: PCA or clustering analysis reveals strong grouping by processing date, laboratory, or reagent lot rather than your biological variables of interest.
Solution: This indicates substantial batch effects requiring computational correction:
- Apply Specialized Correction Algorithms: Use methods specifically designed for methylation data, such as ComBat-met, which employs beta regression to account for the bounded nature of beta-values [80]. Traditional methods like ComBat assume normally distributed data and may perform poorly on raw beta-values.
- Consider Reference-Based Correction: If you have included common reference samples across batches, use ratio-based scaling methods that transform feature values relative to the reference samples [78]. This approach is particularly effective when batch and biological factors are confounded.
- Validate Correction Effectiveness: Re-run visualization after correction to ensure batch separation has been reduced while biological signals have been preserved [79].
Critical Note: Always verify that biological signals of interest have not been removed during batch correction. Over-correction can eliminate genuine biological differences along with technical artifacts [79].
How do I handle newly generated data without reprocessing my entire dataset?
Problem: You need to incorporate new batches of data into an existing corrected dataset but want to avoid completely re-processing all previous data.
Solution: Implement incremental batch correction frameworks:
- Use iComBat for Longitudinal Studies: The newly developed iComBat method extends the traditional ComBat framework to allow adjustment of newly added batches without re-correcting previously processed data [76]. This is particularly valuable for long-term multi-center studies where data collection occurs sequentially over time.
- Maintain Consistent Reference Materials: Include the same reference samples (e.g., control sperm samples) in each new batch to facilitate ratio-based correction approaches that don't require full dataset re-processing [78].
- Preserve Model Parameters: When using empirical Bayes methods like ComBat, save the parameter estimates from your original correction to inform adjustment of new batches [76].
Table: Comparison of Batch Effect Correction Methods for DNA Methylation Data
| Method | Best For | Data Type | Advantages | Limitations |
|---|---|---|---|---|
| ComBat-met [80] | DNA methylation beta-values | Beta-values (0-1 range) | Specifically designed for methylation data; handles bounded distribution | Requires sufficient sample size per batch |
| iComBat [76] | Longitudinal/multi-center studies | M-values | Incremental correction without reprocessing old data | Relatively new method; less established |
| Ratio-based Scaling [78] | Confounded batch-biology scenarios | Normalized counts or values | Effective even when batch completely confounded with biology | Requires reference materials in each batch |
| Harmony [79] | High-dimensional data integration | PCA-reduced data | Efficient for large datasets; preserves fine biological structures | Works on reduced dimensions, not original data |
| ComBat [76] [81] | Balanced batch designs | M-values | Established method; robust to small sample sizes | Assumes normal distribution after transformation |
What are the signs of overcorrection in batch effect adjustment?
Problem: After batch correction, your biological signals seem diminished or unexpected patterns emerge in the data.
Solution: Recognize and address overcorrection:
- Check for Loss of Biological Signal: If known biological differences (e.g., between cell types or treatment conditions) disappear after correction, you may be overcorrecting [79].
- Examine Cluster Markers: After correction, if cluster-specific markers include genes with widespread high expression (e.g., ribosomal genes) rather than specific biological markers, overcorrection may have occurred [79].
- Validate with Positive Controls: Use known biological positives (e.g., imprinted genes with expected methylation patterns in sperm) to ensure they remain detectable after correction.
- Compare Multiple Methods: Apply different correction approaches and compare results. If biological signals vary dramatically between methods, refine your correction strategy.
Prevention: When using algorithms like ComBat, avoid over-shrinking parameters and consider using reference-based correction when possible to preserve biological variability [80].
Experimental Protocols for Batch Effect Management
Standardized Protocol for Multi-Center Sperm Methylation Analysis
This protocol provides a standardized workflow for generating DNA methylation data across multiple laboratories while minimizing batch effects.
Materials Required:
- Sperm samples from all experimental conditions
- Common reference sperm samples (pooled or commercial)
- DNA extraction kits (same lot number across centers)
- Bisulfite conversion kits (same lot number)
- Methylation array or sequencing platform
- Quality control reagents (BioAnalyzer, Qubit)
Procedure:
Experimental Design Phase:
- Implement a balanced design where each center processes samples from all biological groups.
- Include technical replicates of reference samples in each batch.
- Randomize sample processing order to avoid confounding biological groups with processing time.
Sample Preparation:
- Use DNA extraction and bisulfite conversion kits from the same manufacturer and lot number across all centers.
- Standardize DNA quantification methods (use fluorometric methods like Qubit rather than spectrophotometry).
- Establish uniform quality thresholds for DNA integrity (e.g., DIN > 7.0).
Data Generation:
- Process all samples using the same platform (e.g., Illumina EPIC array or specific bisulfite sequencing protocol).
- Include the same control samples in each run to monitor technical variation.
Data Preprocessing:
Batch Effect Assessment:
- Generate PCA plots colored by batch center and biological group.
- Calculate batch effect metrics (e.g., kBET) to quantify effect size.
- Proceed to correction only if significant batch effects are detected.
Batch Effect Correction:
- Apply ComBat-met for methylation array data [80] or ratio-based scaling if reference materials are available [78].
- For longitudinal studies adding new batches, use iComBat to avoid reprocessing existing data [76].
- Validate correction by confirming reduction in batch clustering while preservation of biological signals.
Protocol for Incremental Data Integration in Longitudinal Studies
This protocol addresses the challenge of integrating new data batches without reprocessing previously corrected datasets, which is common in long-term multi-center studies.
Procedure:
Initial Baseline Establishment:
- Process a sufficient number of samples (recommended n≥8 per biological group) to establish a robust baseline.
- Apply standard batch correction (e.g., ComBat-met) to this initial dataset.
- Save all model parameters and distribution estimates from this correction.
Reference Material Inclusion:
- Include the same reference sperm samples in all subsequent batches.
- Ensure reference materials are processed identically to test samples.
New Batch Processing:
- Process new samples alongside reference materials using identical protocols.
- Preprocess new data separately (normalization, background correction).
Incremental Correction:
Quality Assurance:
- Verify that the integrated dataset shows minimal batch separation in PCA.
- Confirm preservation of expected biological patterns using positive controls.
Batch Effect Correction Pathways & Workflows
Decision Framework for Batch Correction Method Selection
Choosing the appropriate batch correction strategy depends on your experimental design, data type, and the specific batch effect challenges you face. The following workflow provides a systematic approach to method selection:
Comprehensive Multi-Center Data Integration Workflow
For complex multi-center epigenetic studies, a comprehensive approach integrating multiple quality control and correction steps is essential:
The Scientist's Toolkit: Essential Research Reagents & Materials
Successful multi-center epigenetic standardization requires careful selection and consistent use of key reagents and materials. The following table details essential components for batch-effect-aware sperm epigenetic studies:
Table: Essential Research Reagents & Materials for Multi-Center Sperm Epigenetic Studies
| Reagent/Material | Function | Standardization Consideration | Batch Effect Relevance |
|---|---|---|---|
| Reference Sperm Samples | Quality control and ratio-based correction | Pooled samples from multiple donors; aliquoted and distributed to all centers | Enables ratio-based scaling; monitors technical variation across batches [78] |
| DNA Extraction Kits | Nucleic acid purification | Same manufacturer and lot number across all centers | Minimizes protocol-specific bias in DNA quality and yield [77] |
| Bisulfite Conversion Kits | Chemical conversion of unmethylated cytosines | Same manufacturer and lot number; standardized incubation conditions | Conversion efficiency varies between kits/lots, major source of batch effects [80] |
| Methylation Standards | Positive controls for methylation levels | Commercially available methylated and unmethylated DNA controls | Verifies assay performance and enables cross-batch normalization [78] |
| Library Preparation Kits | Sequencing library construction | Same manufacturer and lot number for all centers | Different ligation efficiencies can introduce batch-specific biases [82] |
| Quality Control Reagents (BioAnalyzer, Qubit) | Assessment of DNA quality and quantity | Standardized quantification methods across centers | Prevents introduction of biases from inaccurate quantification [82] |
Frequently Asked Questions (FAQs)
Q1: What is the difference between normalization and batch effect correction?
A1: Normalization addresses technical variations between individual samples, such as differences in sequencing depth, library size, or amplification bias. It operates on the raw data matrix and aims to make samples comparable by adjusting for global technical differences. In contrast, batch effect correction specifically addresses systematic differences between groups of samples processed at different times, locations, or conditions. It typically occurs after normalization and focuses on removing batch-specific biases while preserving biological signals [79].
Q2: Can I correct for batch effects if I didn't include reference samples in my experiment?
A2: Yes, but with limitations. Methods like ComBat, ComBat-met, and SVA can estimate and remove batch effects without reference samples by leveraging the entire dataset's structure [76] [80]. However, these methods perform best when biological and batch factors are not completely confounded. When all samples from one biological group are processed in a single batch, distinguishing biological signals from technical artifacts becomes challenging without reference samples [78]. For future studies, always include reference materials.
Q3: How many samples per batch do I need for effective batch correction?
A3: While requirements vary by method, most batch correction algorithms require a minimum of 3-5 samples per batch for stable parameter estimation [76]. For complex designs or when using empirical Bayes methods, 8-10 samples per batch provides more robust correction. For multi-center studies, ensure each center processes sufficient samples from all biological groups to enable accurate batch effect estimation.
Q4: Are batch effects more severe in single-cell epigenetics compared to bulk analyses?
A4: Yes, single-cell epigenetic technologies (e.g., scATAC-seq, single-cell bisulfite sequencing) typically exhibit more pronounced batch effects due to lower input material, higher technical noise, increased dropout rates, and cell-to-cell variability [77] [79]. Correction methods developed for bulk data may be insufficient for single-cell data, requiring specialized approaches like Harmony or Seurat that account for data sparsity and high dimensionality [79].
Q5: What should I do if different batch correction methods give substantially different results?
A5: When methods disagree, follow this systematic approach:
- Verify that all methods were applied appropriately with correct parameter settings.
- Use biological positive controls to determine which result best preserves expected signals.
- Check if your data violates assumptions of any method (e.g., normal distribution for ComBat).
- Consider using ensemble approaches or reporting results from multiple methods.
- Consult recent benchmarking studies to identify methods most suitable for your data type [78] [80].
Q6: Can batch effects be completely eliminated from multi-center epigenetic studies?
A6: Complete elimination is challenging and often undesirable, as over-correction can remove biological signals. The practical goal is to reduce batch effects sufficiently so that they no longer dominate the analytical results or lead to false conclusions [77]. With careful experimental design, appropriate correction methods, and validation, batch effects can be effectively mitigated to enable robust multi-center analyses. The combination of proper study design, standardized protocols, reference materials, and computational correction typically provides the most reliable approach.
Establishing Internal Reference Standards and Controls for Inter-Laboratory Calibration
Frequently Asked Questions (FAQs) and Troubleshooting Guides
1. What are the critical control points in a sperm epigenetics workflow to ensure inter-laboratory reproducibility?
The entire workflow, from sample collection to data analysis, requires standardization. The table below outlines key control points and their associated challenges based on recent research.
Table 1: Critical Control Points in Sperm Epigenetic Analysis
| Workflow Stage | Key Control Point | Potential Challenge | Suggested Control/Metric |
|---|---|---|---|
| Sample Collection & Processing | Sperm Purity | Somatic cell contamination [83] | Use somatic cell lysis buffer; validate with purity markers. |
| Nucleic Acid Isolation | DNA/RNA Integrity | Fragmentation; incomplete bisulfite conversion [83] [6] | Measure DNA integrity number; include conversion controls in assays. |
| Epigenetic Assay | DNA Methylation Measurement | Technical variation in platform and analysis [84] [85] | Use a standardized panel of imprinted genes; implement robust correlation metrics (e.g., Normalized Mutual Information) [85]. |
| Data Analysis & Normalization | Inter-laboratory Data Comparison | Batch effects; different bioinformatic pipelines [85] | Use common reference standards; adopt standardized normalization procedures and quality metrics. |
2. Which specific DNA methylation biomarkers can serve as a core panel for calibrating assays across labs?
Research has identified several imprinted genes whose methylation status is consistently associated with infertility outcomes. Combining these into a multi-gene signature improves diagnostic power and provides a robust set of loci for inter-laboratory calibration.
A 2023 study systematically evaluated combinations of imprinted genes and validated a 5-gene panel for identifying epigenetic abnormalities in sperm. The performance of different combinations is summarized below [83].
Table 2: Performance of Multi-Gene DNA Methylation Biomarker Panels for Identifying Epigenetic Abnormalities
| Number of Genes | Genes in Combination | Area Under the Curve (AUC) | Specificity (%) | Sensitivity (%) |
|---|---|---|---|---|
| 7 | IGF2-H19, IG-DMR, ZAC, KvDMR, PEG3, MEST, PEG10 | 0.89 | 92.65 | 69.12 |
| 6 | IGF2-H19, IG-DMR, ZAC, KvDMR, PEG3, MEST | 0.88 | 90.14 | 73.08 |
| 5 | IGF2-H19, IG-DMR, ZAC, KvDMR, PEG3 | 0.88 | 90.41 | 70.00 |
| 4 | IGF2-H19, IG-DMR, ZAC, KvDMR | 0.78 | 91.03 | 52.74 |
The combination of IGF2-H19 DMR, IG-DMR, ZAC, KvDMR, and PEG3 provides an optimal balance of high specificity and a high AUC, making it a strong candidate for a core calibration panel [83]. Other frequently cited genes in the literature include H19, MEST, and PLAGL1 [84].
3. Our lab is getting poor correlation between technical replicates in our ATAC-seq data. What steps can we take?
Poor correlation in epigenomic assays like ATAC-seq is often due to data characteristics and the choice of correlation metrics. Standard correlation coefficients (e.g., Pearson's R) can be misleading with genomic data that contains many regions with zero signal (co-zeros) [85].
- Troubleshooting Steps:
- Remove Co-Zeros: Filter out genomic bins where both replicates have zero signal before calculating correlation. This prevents artificially high correlation from regions with no accessible chromatin [85].
- Use Appropriate Metrics: Replace Pearson's R with metrics that are better suited for epigenomic data. Normalized Mutual Information (NMI) and the R² coefficient have been shown to provide a more accurate assessment of reproducibility after co-zero removal [85].
- Check Sequencing Depth: Ensure replicates have comparable sequencing depths and FrIP scores (fraction of reads in peaks) as defined by ENCODE standards [85].
- Verify Sample Quality: Re-assess sample quality from the beginning of the workflow, including cell integrity and library preparation.
4. What are the essential reagents and materials required for establishing a standardized sperm epigenetics protocol?
The table below lists key reagents and their functions for core epigenetic analyses in sperm.
Table 3: Research Reagent Solutions for Sperm Epigenetic Analysis
| Reagent/Material | Function | Key Consideration |
|---|---|---|
| Somatic Cell Lysis Buffer [83] | Removes contaminating somatic cells from semen samples prior to DNA extraction, ensuring analysis is specific to sperm. | Critical for obtaining a pure sperm epigenetic profile. |
| Bisulfite Conversion Kit | Converts unmethylated cytosines to uracils, allowing for the quantification of DNA methylation. | Efficiency of conversion must be monitored and reported. |
| Primers for Imprinted Genes | Amplify regions of interest (e.g., from the 5-gene panel) for downstream methylation analysis. | Sequences should be consistent across labs for calibration [83]. |
| Pyrosequencing System | Provides quantitative, base-resolution measurement of DNA methylation levels at specific CpG sites. | A preferred method for validating and quantifying methylation in targeted regions [83]. |
| Antibodies for Histone Modifications | Used in ChIP-seq to map the enrichment of specific histone marks (e.g., H3K4me3, H3K27ac) [86] [37]. | Validation and specificity of antibodies are crucial for reproducibility. |
| Calibrated Reference Sperm Samples | Pooled sperm samples from characterized donors (e.g., fertile vs. infertile) to be used as internal controls in every experiment. | The cornerstone of inter-laboratory calibration. |
Experimental Protocols for Key Assays
Detailed Protocol: DNA Methylation Analysis of Sperm by Pyrosequencing
This protocol is adapted from a 2023 study that established a diagnostic panel for recurrent pregnancy loss [83].
Sample Collection and Somatic Cell Lysis:
- Collect semen samples after 3-5 days of sexual abstinence.
- Remove seminal plasma by centrifugation.
- Treat the sperm pellet with a somatic cell lysis buffer (0.1% SDS, 0.5% Triton X-100) for 6 hours at room temperature on a shaker to lyse any contaminating somatic cells.
- Wash the purified sperm pellet twice with phosphate-buffered saline (PBS) and store at -80°C.
DNA Extraction and Bisulfite Conversion:
- Extract genomic DNA from the purified sperm using a commercial kit designed for sperm DNA purification.
- Subject 500 ng of genomic DNA to bisulfite conversion using a commercial bisulfite conversion kit, following the manufacturer's instructions. This deaminates unmethylated cytosines to uracils, while methylated cytosines remain unchanged.
PCR Amplification:
- Perform PCR amplification on the bisulfite-converted DNA using biotinylated primers specific to the differentially methylated regions (DMRs) of the target genes (e.g., IGF2-H19, IG-DMR, ZAC, KvDMR, PEG3).
- Use a hot-start PCR kit to improve specificity.
Pyrosequencing:
- Bind the biotinylated PCR product to streptavidin-coated sepharose beads.
- Wash and denature the double-stranded DNA to obtain a single-stranded template.
- Load the template into a pyrosequencing cartridge along with the sequencing primer.
- Run the sample on the pyrosequencer. The instrument sequentially dispenses nucleotides, and light is emitted upon incorporation (as pyrophosphate is released), yielding a quantitative readout of the methylation percentage at each CpG site within the amplicon.
Data Analysis:
- Calculate the average methylation level for each gene locus from the individual CpG sites.
- For a multi-gene panel, the methylation values can be combined into a single probability score using multiple logistic regression to classify samples as epigenetically normal or abnormal [83].
Workflow and Relationship Diagrams
Diagram 1: Standardized Sperm Epigenetics Workflow
Diagram 2: Key Sperm Epigenetic Components
From Bench to Bedside: Clinical Validation and Technology Assessment
Troubleshooting Guides & FAQs
Frequently Asked Questions (FAQs)
Q1: What is the core epigenetic mechanism being investigated for male fertility assessment? A: The primary mechanism is DNA methylation, a biochemical process that adds a methyl group to a cytosine nucleotide, typically at CpG dinucleotides. In gene promoters, this modification usually leads to gene silencing. The maintenance of proper DNA methylation patterns is crucial for healthy sperm function and embryonic development. Aberrant methylation in sperm has been consistently linked to impaired spermatogenesis and reproductive dysfunction [87] [88].
Q2: How strong is the evidence linking sperm epigenetic marks to Intrauterine Insemination (IUI) outcomes? A: Evidence is robust and shows a significant association. A large cohort study found that men with an "Excellent" sperm epigenetic profile (≤3 dysregulated promoters) had a live birth rate of 44.8% with IUI, compared to only 19.4% for men with a "Poor" profile (≥22 dysregulated promoters). This difference was statistically significant (P=.03), indicating that high levels of epigenetic instability in sperm are strongly associated with lower IUI success [87].
Q3: Does in vitro fertilization (IVF) overcome epigenetic defects in sperm? A: Yes, evidence suggests that IVF, particularly when performed with intracytoplasmic sperm injection (ICSI), can overcome high levels of sperm epigenetic instability. The same study that found significant outcome differences with IUI showed no significant differences in live birth outcomes among the poor, average, and excellent epigenetic groups when IVF/ICSI was used. This implies that the IVF/ICSI procedure bypasses the functional limitations associated with aberrant sperm epigenetics [87].
Q4: Which specific genes or genomic regions are most critical to analyze? A: Research points to several key areas:
- Imprinted Genes: The H19/IGF2 locus (paternally methylated) and the MEST gene (maternally methylated) have been repeatedly linked to male infertility and embryo development [88].
- Promoters of Developmental Genes: A panel of 1,233 gene promoters with stable methylation in fertile donors was identified as a key biomarker. Dysregulation in these promoters correlates with sperm quality and IUI outcomes [87].
- Repetitive Elements: Proper methylation of retrotransposons, like LINE1, is essential. Lack of methylation can lead to genomic instability and has been associated with male infertility [88].
Q5: My lab is new to this field. What is the basic workflow for analyzing sperm DNA methylation? A: A generalized core workflow is as follows. Specific protocols will vary based on the chosen technology [87] [89].
Troubleshooting Common Experimental Issues
Issue: High background noise in methylation data from sperm samples. Solution: A critical first step is to ensure your sample is free of somatic cell contamination. Somatic cells have vastly different methylation profiles that can confound results.
- Action: Before analysis, filter out samples that do not meet a sperm-specific methylation threshold. One validated method is to check the mean methylation value of probes in the differentially methylated region of the DLK1 gene. Samples with a mean value ≥0.24 in this region may contain significant somatic cell DNA and should be excluded [87].
Issue: Inconsistent results when trying to replicate published epigenetic biomarker panels. Solution: Inconsistent replication often stems from a lack of standardized data analysis and classification criteria.
- Action: Adopt a standardized framework for defining "dysregulation." Establish your methylation variability cutoffs based on a reference set of fertile donor samples. When analyzing samples from men with infertility, count the number of promoters that fall outside these fertile-derived cutoffs. Classify samples into categories (e.g., Poor, Average, Excellent) based on the number of dysregulated promoters, using consistent percentile thresholds (e.g., top and bottom 10th percentiles) [87].
Issue: My study has insufficient statistical power to detect significant associations. Solution: This is a common challenge in epigenetic association studies.
- Action: Conduct a power analysis before beginning your study. For validation studies, it is highly recommended to include both a discovery and an independent validation cohort. Ensure your sample size is large enough to detect realistic effect sizes after adjusting for multiple testing. Account for potential confounders, such as cell-type heterogeneity and key clinical variables (e.g., female partner's age and fertility status) [89].
Table 1: Sperm Epigenetic Quality and its Correlation with Clinical Outcomes After IUI (Cumulative over 2-3 cycles) [87]
| Sperm Epigenetic Quality Group | Number of Dysregulated Promoters | Pregnancy Rate | Live Birth Rate |
|---|---|---|---|
| Excellent | ≤ 3 | 51.7% | 44.8% |
| Average | 4 - 21 | Not specified | Significantly higher than Poor group |
| Poor | ≥ 22 | 19.4% | 19.4% |
Table 2: Key Imprinted Genes Recurrently Associated with Male Infertility and Altered Sperm Methylation [88]
| Gene | Genomic Imprint Status | Functional Role | Association with Infertility |
|---|---|---|---|
| H19 | Maternally expressed (paternally methylated) | Codes for a non-coding RNA; regulates growth. | Hypomethylation in sperm is linked to infertility. |
| IGF2 | Paternally expressed (maternally methylated) | Fetal growth factor. | Often found with aberrant methylation in infertile men. |
| MEST (PEG1) | Paternally expressed (maternally methylated) | Involved in embryonic development. | Hypermethylation in sperm is associated with poor semen quality. |
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Research Reagent Solutions for Sperm Epigenetic Analysis
| Item | Function in Protocol | Key Considerations |
|---|---|---|
| Sperm Purification Kits | Isolate pure spermatozoa from semen; critical for removing contaminating somatic cells. | Purity is paramount. Validate with a somatic cell marker (e.g., DLK1 DMR methylation) [87]. |
| Bisulfite Conversion Kit | Chemically converts unmethylated cytosines to uracils, while methylated cytosines remain unchanged. | The cornerstone of most methylation assays. Efficiency of conversion must be verified [89]. |
| Infinium MethylationEPIC BeadChip | Genome-wide microarray for analyzing methylation at >850,000 CpG sites. | Ideal for discovery-phase studies. Covers promoters, enhancers, and gene bodies [87]. |
| Pyrosequencing Platform | Quantitative, locus-specific method for analyzing methylation at a small number of CpG sites. | Excellent for validating findings from genome-wide screens or for focused clinical assays [89] [90]. |
| Antibodies for Histone Modifications (e.g., H3K4me3) | Used in ChIP-seq to map the genomic location of specific histone modifications in sperm. | Relevant for investigating the paternal chromatin landscape's role in development [37]. |
Standardized Experimental Protocol: Sperm DNA Methylation Analysis via Microarray
This protocol outlines the key steps for a genome-wide sperm DNA methylation analysis using the Illumina Infinium MethylationEPIC array, based on methodologies from published studies [87].
Sample Collection and Sperm Purification
- Collect semen samples following standard clinical procedures.
- Purify sperm cells using a density gradient centrifugation method to minimize contamination from seminal plasma and somatic cells (e.g., leukocytes). This step is critical for data integrity.
DNA Extraction and Quality Control
- Extract genomic DNA from purified sperm using a commercial kit designed for mammalian cells.
- Quantify DNA using a fluorometer and assess purity via spectrophotometry (260/280 ratio ~1.8).
Bisulfite Conversion and Microarray Processing
- Subject 500 ng of genomic DNA to sodium bisulfite conversion using a dedicated kit (e.g., Zymo EZ DNA Methylation Kit). This reaction deaminates unmethylated cytosines to uracils.
- Perform the Illumina Infinium MethylationEPIC assay according to the manufacturer's instructions. This includes:
- Amplifying the bisulfite-converted DNA.
- Fragmenting the amplified product.
- Hybridizing the DNA to the EPIC BeadChip.
- Staining and scanning the array to generate intensity data.
Data Pre-processing and Normalization
- Process the raw intensity data (IDAT files) using a standard pipeline like
minfiin R. - Perform background correction and normalization (e.g., functional normalization) to remove technical variation.
- Filter out probes that are known to have common single-nucleotide polymorphisms (SNPs), are cross-reactive, or have a low detection p-value (p > 0.01).
Somatic Cell Contamination Check
- Calculate the mean methylation beta value for all CpG probes within the Differentially Methylated Region (DMR) of the DLK1 gene (chr14:101,191,893–101,192,913, GRCh37).
- Exclude samples with a mean DLK1 DMR methylation value ≥ 0.24, as this indicates potential somatic cell contamination [87].
Analysis of Promoter Dysregulation
- Define a reference set: Identify 1,233 gene promoters that show the least variable methylation in a control group of fertile sperm donors.
- Set variability cutoffs: Establish methylation variability thresholds for each of these promoters based on the fertile control data.
- Analyze patient samples: For each infertility cohort sample, count the number of the 1,233 promoters where methylation falls outside the fertile-derived cutoffs. This count is the "number of dysregulated promoters."
- Classify samples: Categorize samples into groups (e.g., Poor, Average, Excellent) based on the percentile ranks of their dysregulated promoter count [87].
Statistical Correlation with Outcomes
- Use statistical tests (e.g., t-tests, regression models) to compare pregnancy and live birth rates across the different epigenetic quality groups.
- Control for female factors in the analysis by, for example, restricting the cohort to cycles where the female partner is under 35 and has no known infertility diagnoses [87].
The following diagram illustrates the logical pathway from sperm epigenetic state to clinical outcome, summarizing the key concepts discussed.
Core Validation Framework (V3)
For any novel epigenetic test, a foundational three-component framework known as V3 is widely recommended to determine if the test is "fit-for-purpose." This framework is adapted from established practices in digital medicine and biomarker development and is directly applicable to epigenetic assays [91].
The V3 Framework Components [91]:
- Verification: A systematic, bench-level evaluation of the technology. For an epigenetic test, this confirms that the laboratory instrumentation (e.g., sequencer, array scanner) and basic reagents perform to specification.
- Analytical Validation: This step evaluates the data processing algorithms and the assay's performance in measuring the target analyte. It answers the question: "Does the test accurately and reliably measure the specific epigenetic marker (e.g., DNA methylation at a CpG site)?"
- Clinical Validation: This demonstrates that the test's output acceptably identifies, measures, or predicts a relevant clinical, biological, or functional state in the intended patient population.
FAQs: Implementing Validation in Sperm Epigenetics
FAQ 1: What are the unique challenges in validating epigenetic tests for sperm, and how do we address them?
Sperm epigenetic analyses face specific hurdles that must be accounted for in a validation plan [92].
- Challenge: Low DNA Quantity & Quality: Sperm cells contain picogram levels of DNA, and the chromatin is highly compacted. This can lead to amplification bias and increased technical noise.
- Troubleshooting Guide:
- Issue: High sample failure rate or inconsistent results.
- Step 1: Implement a rigorous DNA quantification step (e.g., fluorometry) post-extraction and set a minimum input mass threshold.
- Step 2: Use a robust whole-genome amplification kit validated for bisulfite-converted DNA and include positive control samples in every run.
- Step 3: Standardize the bisulfite conversion protocol across all laboratory sites and track conversion efficiency (>99% is ideal) as a key quality control metric [92].
FAQ 2: How do we define and validate the clinical utility of a sperm epigenetic biomarker?
Clinical validation must prove the test predicts a meaningful outcome. In sperm epigenetics, this could be fertility status or success in assisted reproductive technology (ART) [93] [91].
- Troubleshooting Guide:
- Issue: A sperm DNA methylation signature is statistically different between fertile and infertile cohorts but fails to predict pregnancy outcomes in a clinic.
- Step 1: Ensure the clinical validation study is prospectively designed with a pre-specified primary endpoint (e.g., live birth rate, fertilization rate).
- Step 2: Define and lock the predictive algorithm before conducting the clinical validation study to avoid overfitting.
- Step 3: The study population must be well-characterized and representative of the intended-use population. For example, when validating a test for predicting sperm retrieval success in men with non-obstructive azoospermia (NOA), the cohort should be exclusively composed of NOA patients [94].
FAQ 3: Our lab is getting inconsistent results with the same sample across different runs. How can we improve analytical precision?
Inconsistency often stems from uncontrolled pre-analytical and analytical variables [92].
- Troubleshooting Guide:
- Issue: High intra-sample coefficient of variation (CV) for methylation levels at specific CpG sites.
- Step 1: Audit Pre-analytical Factors: Standardize semen processing, sperm purification method, and DNA extraction protocol across all lab sites. Document any deviations.
- Step 2: Control for Batch Effects: Use a balanced experimental design where cases and controls are processed in the same sequencing or array batch. Employ statistical methods like ComBat to correct for residual technical batch effects discovered during analysis [95].
- Step 3: Establish QC Thresholds: Define acceptable ranges for key metrics (e.g., bisulfite conversion efficiency, read depth, probe signal intensity) and automatically flag samples that fall outside these ranges for re-processing.
Performance Metrics & Benchmarking
For any epigenetic test, key analytical performance metrics must be established and documented. The table below summarizes the core metrics that should be evaluated during analytical validation.
Table 1: Key Analytical Performance Metrics for Epigenetic Tests
| Metric | Definition | Target for Sperm Epigenetic Tests | How to Measure |
|---|---|---|---|
| Precision (Repeatability) | Agreement between replicate measurements of the same sample under identical conditions [92]. | CV < 5% for methylation beta values at target CpGs [92]. | Run the same sample in triplicate in a single batch. Calculate CV for each CpG site. |
| Precision (Reproducibility) | Agreement between measurements of the same sample under changing conditions (e.g., different days, operators, instruments) [92]. | CV < 10% for methylation beta values at target CpGs [92]. | Run the same sample across multiple batches, operators, or sites. |
| Accuracy | Agreement between the test result and an accepted reference standard or truth [92]. | Mean absolute error < 2% against a validated platform (e.g., pyrosequencing) [95]. | Compare results from the novel test to a "gold standard" method on a set of reference samples. |
| Analytical Sensitivity | Lowest quantity of input DNA or lowest level of methylation change the test can reliably detect [92]. | Detect methylation differences of 10% with as low as 10ng input DNA [92]. | Perform a dilution series of methylated and unmethylated DNA controls. |
| Analytical Specificity | Ability to correctly detect the target methylation signal without interference from related but distinct epigenetic marks or genomic variations. | >99% specificity in a background of fragmented DNA [96]. | Spike-in known contaminants (e.g., non-sperm DNA, heme) and confirm target signal is unchanged. |
When benchmarking your test against public datasets or other models, it is critical to use standardized approaches. For example, in epigenetic age assessment, the EnsembleAge framework uses multiple models to improve robustness, a concept that can be applied to sperm epigenetics to harmonize results across different laboratories and platforms [97].
Table 2: Example DNA Methylation Detection Techniques for Sperm Epigenetics
| Technique | Key Feature | Best Use Case in Sperm Research | Throughput |
|---|---|---|---|
| Whole-Genome Bisulfite Sequencing (WGBS) | Single-base resolution genome-wide methylation mapping [95]. | Discovery of novel differentially methylated regions (DMRs) between fertile and infertile men. | Low |
| Illumina Infinium Methylation BeadChip | Interrogates predefined CpG sites; cost-effective and reproducible [95]. | Large-scale cohort studies and validation of DMRs from WGBS. | High |
| Reduced Representation Bisulfite Sequencing (RRBS) | Sequences CpG-rich regions; balances cost and coverage [95]. | Targeted discovery when WGBS is too expensive. | Medium |
| Methylation-Specific PCR (MSP) | Detects methylation at a specific, short genomic region [95]. | Rapid, low-cost validation of a single DMR of high interest in many samples. | High |
The Scientist's Toolkit: Research Reagent Solutions
Standardizing reagents across laboratories is fundamental to generating comparable data. Below is a list of essential materials and their functions for sperm epigenetic studies.
Table 3: Essential Research Reagents for Sperm Epigenetic Protocols
| Reagent / Material | Function | Considerations for Standardization |
|---|---|---|
| Sperm Washing Buffer | To remove seminal plasma and non-sperm cells without damaging spermatozoa. | Standardize buffer composition (e.g., PBS vs. specialized commercial buffers) and centrifugation speed/time across labs. |
| DNA Extraction Kit | To isolate high-quality, high-molecular-weight genomic DNA from purified sperm. | All participating labs should use the same kit and protocol, validated for sperm cells. |
| Bisulfite Conversion Kit | To convert unmethylated cytosines to uracils, while leaving methylated cytosines unchanged. | This is a critical step. Use the same kit and strictly control incubation temperature and time. |
| DNA Methylation Standard | A control with known methylation levels at specific loci (e.g., 0%, 50%, 100% methylated DNA). | Include these in every batch to monitor bisulfite conversion efficiency and assay performance. |
| Library Prep Kit | For preparing sequencing libraries from bisulfite-converted DNA. | Standardizing the kit minimizes bias introduced during library amplification. |
| Methylation Array | The specific platform (e.g., EPIC) used for genome-wide methylation profiling. | Ensure all labs use the same array version and processing chip type. |
Technical Support Center: Troubleshooting Sperm Epigenetic Analysis
Frequently Asked Questions (FAQs)
1. FAQ: My chromatin preparation from sperm tissue yields very low DNA concentration. What could be the cause and how can I improve it?
- Problem: Low DNA concentration after chromatin fragmentation from sperm cells.
- Detailed Description: When harvesting cross-linked chromatin from tissue samples, the yield of chromatin can vary significantly between tissue types. Sperm cells, with their highly compacted chromatin structure, can present particular challenges. A low concentration can lead to failed subsequent steps like immunoprecipitation or sequencing library preparation [98].
- Suggested Remedies:
- Verify Input Material: Ensure you are starting with an adequate number of sperm cells. The issue may stem from an inaccurate initial cell count [98].
- Assess Lysis Efficiency: If using an enzymatic protocol, visualize cell nuclei under a microscope before and after sonication to confirm complete lysis of nuclei. Incomplete lysis will result in poor chromatin recovery [98].
- Increase Input: If the DNA concentration is close to but below the required threshold (e.g., 50 µg/ml), you can add a larger volume of chromatin to each immunoprecipitation reaction to reach the recommended amount (e.g., 5–10 µg) [98].
- Tissue-Specific Consideration: Be aware that different tissues have inherently different chromatin yields. For instance, brain and heart tissue typically yield much less chromatin (2–5 µg per 25 mg of tissue) compared to spleen or liver [98]. While this data is not sperm-specific, it highlights the importance of optimizing for your specific cell type.
2. FAQ: My DNA methylation data from sperm samples shows high background noise or low resolution. Should I switch from traditional bisulfite sequencing to an emerging method?
- Problem: Bisulfite sequencing, the traditional gold standard for DNA methylation (5mC) detection, can lead to degraded DNA and high background due to severe DNA damage from the bisulfite conversion process. This is particularly problematic with precious sperm samples [99].
- Detailed Description: Bisulfite treatment deaminates unmodified cytosines to uracils but leaves 5mC and 5hmC intact. However, this process fragments and damages DNA, requiring high input amounts and potentially introducing biases [99].
- Suggested Remedies & Comparative Analysis:
- Traditional Method (WGBS): Whole-genome bisulfite sequencing is base-resolution and quantitative but suffers from the drawbacks mentioned above [99].
- Emerging Automated/Novel Platforms: Consider newer, non-bisulfite methods that are becoming more accessible for standardized protocols.
- EM-Seq (Enzymatic Methyl Sequencing): Uses enzymes instead of bisulfite to identify methylated cytosines, resulting in significantly less DNA damage and lower input requirements [99].
- TAPS (TET-Assisted Pyridine Borane Sequencing): A chemical-assisted method that is non-destructive to DNA and can be more cost-effective [99].
- Recommendation: For standardizing protocols across labs, especially when sample integrity and consistency are paramount, transitioning from WGBS to EM-Seq is a highly recommended strategy to reduce technical variability and improve data quality [99].
3. FAQ: When profiling histone modifications in sperm, I get inconsistent results between replicates. How can I standardize this process?
- Problem: Inconsistent ChIP-Seq results for histone marks in sperm samples, likely due to the technical variability of the cross-linking and immunoprecipitation steps [99].
- Detailed Description: The classical ChIP-Seq method involves formaldehyde cross-linking, which can cause false positives and requires large input DNA amounts. Antibody quality and specificity are also major sources of variability between experiments and laboratories [99].
- Suggested Remedies & Comparative Analysis:
- Traditional Method (ChIP-Seq): The well-established but highly variable standard. Its limitations include epitope masking from cross-linking and high background noise [99].
- Emerging Automated/Novel Platforms: Newer, in-situ methods offer a path to greater standardization.
- CUT&RUN (Cleavage Under Targets and Release Using Nuclease): This method immobilizes cells on magnetic beads and uses antibody-guided MNase cleavage to release target protein-DNA complexes. It operates at high resolution (~20 bp) and has a much lower background than ChIP-Seq because it avoids cross-linking [99].
- CUT&Tag (Cleavage Under Targets and Tagmentation): An improvement on CUT&RUN that uses a protein A-Tn5 transposase fusion protein to simultaneously cleave and tag the target chromatin for sequencing. This simplifies library construction, reduces hands-on time, and is amenable to automation and high-throughput workflows, making it ideal for standardization across labs [99].
- Recommendation: For profiling histone modifications in sperm, adopting CUT&Tag is strongly advised for protocol standardization. It requires fewer cells, has higher reproducibility, and its simpler workflow is less prone to technical artifacts [99].
4. FAQ: I am investigating transgenerational epigenetic inheritance through the male germline. What are the key epigenetic factors in sperm I should focus on?
- Problem: The molecular mechanisms underlying the transmission of epigenetic information through sperm are complex and not fully understood, making experimental design challenging [57] [59].
- Detailed Description: Sperm were once considered mere DNA carriers, but they contain a complex payload of epigenetic information. This includes not just protamine-packaged DNA, but also nucleosome-retained regions, various RNA species, and DNA methylation marks that can influence embryonic development and offspring phenotype [57].
- Suggested Remedies & Research Focus:
- DNA Methylation: This is the most studied epigenetic marker. Focus on imprinted genes (e.g., H19, MEST, SNRPN), as disruptions in their methylation are strongly linked to male infertility and could be a mechanism for inheritance [43].
- Sperm Chromatin (Histone Modifications): Approximately 4–15% of the human sperm genome retains nucleosomes, and these are enriched at key developmental gene promoters (e.g., HOX genes) [57]. Profiling histone marks like H3K4me2/3 and H3K27ac at these regions is crucial.
- Sperm RNA Payload: Sperm are transcriptionally inactive but contain a rich repertoire of fragmented mRNAs and small non-coding RNAs (e.g., miRNAs, piRNAs, tRNA fragments) [57]. These RNAs are a record of spermatogenesis and have been shown to influence preimplantation development and offspring metabolic and behavioral responses [57].
- Recommendation: A multi-faceted approach is necessary. Do not focus on a single epigenetic layer. Standardized protocols should enable parallel analysis of DNA methylation (using EM-Seq), histone modifications (using CUT&Tag), and the sperm RNA transcriptome to build a comprehensive picture [57] [59].
Comparative Data Analysis Tables
Table 1: Comparison of Key Technologies for Sperm Epigenetic Analysis
| Feature | Traditional Method (ChIP-Seq / WGBS) | Emerging Platform (CUT&Tag / EM-Seq) |
|---|---|---|
| Key Principle | Cross-linking & antibody enrichment (ChIP-Seq); Bisulfite conversion (WGBS) [99] | In-situ cleavage & tagmentation (CUT&Tag); Enzymatic conversion (EM-Seq) [99] |
| Resolution | ~200 bp (ChIP-Seq); Base (WGBS) [99] | ~20 bp (CUT&Tag); Base (EM-Seq) [99] |
| Input Material | High (1-5 million cells) [99] | Low (<100,000 cells) [99] |
| DNA Damage | High (WGBS) [99] | Low [99] |
| Background Noise | High (ChIP-Seq) [99] | Low [99] |
| Protocol Simplicity | Complex, multi-day [99] | Simplified, faster [99] |
| Ease of Standardization | Low (high technical variability) [99] | High (more streamlined workflow) [99] |
| Best for Sperm Analysis | When large sample quantities are available | For precious low-count samples and multi-lab standardization |
Table 2: Expected Chromatin Yield from Different Tissues (for Troubleshooting) [98]
| Tissue Type | Total Chromatin Yield (per 25 mg tissue) |
|---|---|
| Spleen | 20–30 µg |
| Liver | 10–15 µg |
| Kidney | 8–10 µg |
| Brain | 2–5 µg |
| Heart | 2–5 µg |
| HeLa Cells | 10–15 µg (per 4 x 10^6 cells) |
Note: Sperm cells are not listed in this reference data, but this table provides a critical benchmark. If your sperm chromatin yields are significantly lower than the range for brain/heart tissue, it indicates a problem with the isolation or fragmentation protocol that needs optimization [98].
Experimental Workflow Visualization
Sperm Epigenetic Analysis Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Sperm Epigenetics Research
| Reagent / Material | Function in Experiment |
|---|---|
| Micrococcal Nuclease (MNase) | Enzymatically digests and fragments chromatin for analyses like ChIP-Seq or nucleosome positioning studies [98]. |
| Formaldehyde / Paraformaldehyde | Cross-links proteins (histones, transcription factors) to DNA, preserving in vivo interactions for ChIP-Seq protocols [99]. |
| Protein A-MNase / Protein A-Tn5 | Key enzymes for emerging platforms. Protein A-MNase is used in CUT&RUN, and the Protein A-Tn5 fusion protein is the core of the CUT&Tag technology for targeted tagmentation [99]. |
| S-Adenosyl Methionine (SAM) | The universal methyl donor for methylation reactions, used in studies involving DNA methyltransferases (DNMTs) [100]. |
| Specific Antibodies | Critical for any enrichment-based method (ChIP-Seq, CUT&RUN, CUT&Tag). Target-specificity is paramount for success (e.g., anti-H3K4me3, anti-5mC) [99] [43]. |
| Sodium Bisulfite | The key chemical in traditional DNA methylation analysis (WGBS) that converts unmethylated cytosine to uracil [99]. |
| DNMT / TET Inhibitors | Chemical tools (e.g., 5-azacytidine) to manipulate the epigenome and study the functional role of DNA (de)methylation in sperm function [99] [43]. |
The traditional analysis of male fertility has relied on standard semen parameters—concentration, motility, and morphology. However, these criteria offer limited insight into sperm functionality and poorly predict natural fertility or assisted reproductive technology outcomes [101]. In recent years, sperm epigenetic biomarkers have emerged as powerful diagnostic tools that provide a deeper understanding of male factor infertility. Epigenetics encompasses molecular factors around DNA that regulate germline activity independent of DNA sequence, with DNA methylation being one of the most studied mechanisms in sperm [2].
This technical resource examines successful clinical applications of sperm epigenetic biomarkers through detailed case studies, providing standardized protocols and troubleshooting guidance to support implementation across research laboratories. The growing body of evidence demonstrates that epigenetic signatures in sperm can identify underlying causes of idiopathic infertility, predict treatment responsiveness, and offer insights into transgenerational health implications [102] [24].
Clinical Case Studies
DNA Methylation Biomarkers for Recurrent Pregnancy Loss
Background and Clinical Challenge Recurrent Pregnancy Loss affects approximately 1-2% of women globally, with nearly 50% of cases being idiopathic despite extensive female-factor investigation. This highlighted the need for better paternal-factor diagnostics [83].
Experimental Approach and Biomarker Identification Researchers conducted a case-control study comparing sperm DNA methylation patterns in male partners of RPL couples versus fertile controls. Using pyrosequencing, they analyzed differentially methylated regions of imprinted genes and applied multiple logistic regression to develop a diagnostic probability score [83].
Table 1: Diagnostic Performance of the 5-Gene RPL Signature
| Parameter | Value |
|---|---|
| Genes in Signature | IGF2-H19 DMR, IG-DMR, ZAC, KvDMR, PEG3 |
| Area Under Curve (AUC) | 0.88 |
| Threshold Probability Score | 0.61 |
| Specificity | 90.41% |
| Sensitivity | 70% |
| Validation Cohort | 38 control + 45 RPL samples |
Clinical Application and Workflow The established probability score correctly classified 97% of control samples, while identifying 40% of RPL samples as epigenetically abnormal. This signature provides a specific diagnostic tool to identify couples at risk and guide appropriate counseling [83].
Epigenetic Biomarkers for FSH Therapeutic Responsiveness
Background and Clinical Challenge Follicle-stimulating hormone therapy represents a promising treatment for idiopathic male infertility, but patient responsiveness varies significantly. Predicting which patients will benefit remains challenging [6].
Experimental Approach and Biomarker Identification Researchers performed genome-wide DNA methylation analysis using MeDIP-seq on sperm samples from fertile controls and infertile men before and after FSH treatment. They identified distinct differential methylated regions between FSH responders and non-responders [6].
Table 2: FSH Responsiveness Epigenetic Signature
| Parameter | Findings |
|---|---|
| Analytical Method | MeDIP-Seq (genome-wide) |
| DMRs Identified | 56 significant DMRs (p < 1e-05) |
| Key Feature | No overlap with general infertility DMRs |
| Biological Processes | Transcription, signaling, metabolism |
| Clinical Utility | Identifies FSH therapy responders |
Clinical Application The 56 DMR signature enables clinicians to identify patients most likely to benefit from FSH treatment before initiation, personalizing therapeutic strategies and improving outcomes while reducing unnecessary treatment for non-responders [6].
Sperm Epigenetic Biomarkers in Livestock Models
Background and Clinical Challenge Human fertility studies face challenges in controlling for confounding factors. The bull model provides an excellent alternative due to extensive artificial insemination records and controlled breeding conditions [103].
Experimental Approach and Biomarker Identification Researchers analyzed 100 sperm samples from bulls with precisely documented fertility using reduced representation bisulfite sequencing. They identified 490 fertility-related differentially methylated cytosines, most hypermethylated in subfertile bulls [103].
Predictive Model Development and Validation
- A Random Forest model built on the training set (n=67) showed 72% predictive accuracy on the testing set (n=33)
- Validation on an independent cohort of 20 bulls confirmed 72% accuracy on individual ejaculates
- 46 genes targeted by DMCs are involved in embryonic development, sperm function, and maturation
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for Sperm Epigenetic Analysis
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Sperm Processing | Somatic Cell Lysis Buffer (0.1% SDS, 0.5% Triton X-100) [30], Density gradient media (e.g., Isolate Sperm Separation Medium) [101] | Remove somatic cell contamination, isolate motile sperm population |
| DNA Methylation Analysis | Bisulfite conversion kits (e.g., MethylCode Bisulfite Conversion Kit) [83], Pyrosequencing systems (PyroMark Q96 ID) [83], RRBS or MeDIP-seq reagents [6] [103] | Convert unmethylated cytosines to uracils, quantify methylation levels, genome-wide methylation profiling |
| PCR & Sequencing | PyroMark PCR Amplification Kit [83], Primers for imprinted genes (IGF2-H19, IG-DMR, ZACN, etc.) [83], Illumina EPIC arrays [30] | Target amplification, methylation-specific sequencing, array-based methylation screening |
| Data Analysis | Methylation analysis software (e.g., for Random Forest modeling) [103], Somatic contamination assessment tools [30] | Identify DMRs, build predictive models, quality control |
Troubleshooting Guide: FAQs for Sperm Epigenetic Analysis
FAQ 1: How can I minimize somatic cell contamination in sperm samples?
Challenge: Somatic cell contamination significantly skews sperm-specific epigenetic results, particularly in oligozoospermic samples [30].
Solution: Implement a comprehensive contamination control protocol:
- Initial Processing: Wash semen samples with 1X PBS by centrifugation at 200× g for 15 minutes at 4°C [30]
- Microscopic Examination: Inspect samples under microscope (20X objective) to identify somatic cell contamination levels [30]
- Somatic Cell Lysis: Treat with Somatic Cell Lysis Buffer (0.1% SDS, 0.5% Triton X-100 in ddH2O) for 30 minutes at 4°C [83] [30]
- Repeat Inspection: Re-examine under microscope; repeat SCLB treatment if somatic cells persist [30]
- Epigenetic Verification: Assess known somatic-hypermethylated CpG sites (9,564 identified markers available) to confirm purity [30]
FAQ 2: What statistical approaches best validate epigenetic biomarkers?
Challenge: Ensuring identified epigenetic signatures have robust predictive value.
Solution: Apply multiple validation strategies:
- Multiple Logistic Regression: Combine methylation levels of multiple genes into a probability score for sample classification [83]
- ROC Analysis: Determine optimal threshold values balancing sensitivity and specificity [83]
- Random Forest Modeling: Build predictive models using methylation values at significant DMCs [103]
- Independent Cohort Validation: Test predictive models on completely independent sample sets [103]
FAQ 3: How do I handle inter-individual variability in sperm epigenetics?
Challenge: High biological variability between individuals can obscure meaningful epigenetic signatures.
Solution: Implement strict study design controls:
- Precise Phenotyping: Use stringent, well-defined fertility criteria (e.g., corrected non-return rates in bulls) [103]
- Age Matching: Control for age effects by using subjects in narrow age ranges [103]
- Pooling Strategy: Pool multiple ejaculates per individual to minimize physiologically-driven variations [103]
- Sample Size Considerations: Ensure adequate power through appropriate sample sizes; one study used 100 samples for biomarker discovery [103]
FAQ 4: Which genomic regions show most promise as epigenetic biomarkers?
Challenge: Determining which genomic features provide the most reliable epigenetic biomarkers.
Solution: Focus on these validated regions:
- Imprinted Gene Regions: IGF2-H19, IG-DMR, ZAC, KvDMR, and PEG3 show strong diagnostic potential for RPL [83]
- Developmental Gene Promoters: Regions regulating embryonic development and spermatogenesis [103]
- Non-CpG Islands: MeDIP-seq examines 95% of genome comprising low-density CpG regions, providing different information than CpG island-focused arrays [6]
Standardized Experimental Protocols
Protocol: Sperm DNA Methylation Analysis via Pyrosequencing
Application: Targeted analysis of candidate gene methylation, particularly imprinted genes.
Step-by-Step Workflow:
- Sperm Processing and DNA Extraction
- Collect semen samples after 3-5 days of sexual abstinence [83]
- Remove seminal plasma and treat sperm pellet with somatic cell lysis buffer (0.1% SDS, 0.5% Triton X-100) for 6 hours at room temperature on shaker [83]
- Wash twice with PBS and store at -80°C until DNA extraction [83]
- Extract genomic DNA using commercial sperm DNA purification kits [83]
Bisulfite Conversion
- Use 500ng-1μg genomic DNA for bisulfite conversion
- Apply commercial bisulfite conversion kit per manufacturer's instructions [83]
- Elute converted DNA in 20-40μL elution buffer
PCR Amplification and Pyrosequencing
Protocol: Genome-Wide Sperm DNA Methylation Analysis
Application: Discovery-based approaches for identifying novel epigenetic biomarkers.
Method Options:
- Reduced Representation Bisulfite Sequencing (RRBS): Cost-effective method providing nucleotide-level resolution of CpG-rich regions [103]
- Methylated DNA Immunoprecipitation Sequencing (MeDIP-seq): Examines 95% of genome comprising low-density CpG regions [6]
- Whole-Genome Bisulfite Sequencing (WGBS): Comprehensive single-base resolution methylation profiling [24]
Standardized Analysis Pipeline:
- Quality Control: Assess DNA quality and concentration
- Library Preparation: Follow method-specific protocols (RRBS, MeDIP, or WGBS)
- Sequencing: Perform on appropriate platform (Illumina recommended)
- Bioinformatic Analysis:
- Map reads to reference genome
- Calculate methylation percentages at each CpG
- Identify differentially methylated regions
- Perform functional enrichment analysis of DMR-associated genes
- Validation: Confirm key findings with targeted methods (pyrosequencing)
The case studies presented demonstrate the robust potential of sperm epigenetic biomarkers to revolutionize male fertility assessment. From identifying causes of recurrent pregnancy loss to predicting therapeutic responsiveness, these biomarkers provide clinically actionable information beyond standard semen parameters. The standardized protocols and troubleshooting guides provided here offer researchers a foundation for implementing these analyses across laboratories, supporting the crucial goal of protocol standardization in sperm epigenetics.
As research progresses, the integration of epigenetic biomarkers into clinical andrology workflows promises to personalize infertility treatments, improve assisted reproduction outcomes, and provide insights into the transgenerational impacts of paternal health. Continuing refinement of these biomarkers through rigorous validation studies will further establish their role in the clinical evaluation of male factor infertility.
Developing Consensus Guidelines for Reporting Sperm Epigenetic Data
Troubleshooting Common Experimental Challenges
How can I confirm that my sperm DNA methylation data is not contaminated by somatic cells?
Somatic cell contamination is a major confounder in sperm epigenetic studies, as somatic cells have distinctly different methylation profiles. Even low-level contamination can significantly skew results [30].
Solution: Implement a multi-step quality control protocol:
- Microscopic Examination: Visually inspect the semen sample after washing with 1X PBS to identify and count somatic cells [30].
- Somatic Cell Lysis Buffer (SCLB) Treatment: Incubate the sample with a freshly prepared SCLC (e.g., 0.1% SDS, 0.5% Triton X-100 in ddH2O) for 30 minutes at 4°C. Repeat the treatment and microscopic inspection until no somatic cells are detected [30].
- Biomarker Verification: Analyze known CpG sites that are highly methylated in somatic cells but hypomethylated in sperm. A published set of 9,564 CpG sites can serve as markers for contamination [30].
- Data Analysis Cut-off: Apply a conservative methylation difference cut-off (e.g., ≥15%) during bioinformatic analysis to minimize the influence of any residual, undetected contamination [30].
Why might my sperm histone modification data be inconsistent?
The unique and highly compacted nature of sperm chromatin makes the analysis of retained histones technically challenging. Inconsistencies often arise from sample purity and the specificity of antibodies used [37].
Solution:
- Ensure Sample Purity: Rigorously remove somatic cells using the protocol above, as their histone marks will differ from those of sperm [30].
- Validate Antibody Specificity: Use antibodies specifically validated for ChIP-seq or immunofluorescence in sperm cells. Sperm histones contain testis-specific variants (e.g., TH2B, H2A.L.2), and standard antibodies may not bind effectively [37].
- Standardize Protamine Removal: The tight packaging of DNA by protamines can limit antibody access. Ensure protocols include sufficient chromatin decondensation steps while avoiding over-digestion [104].
How do I determine if an epigenetic change is functionally significant for embryo development?
Linking a specific sperm epigenetic alteration to an offspring phenotype is complex due to the extensive epigenetic reprogramming that occurs after fertilization [24].
Solution:
- Focus on Resistant Regions: Prioritize the analysis of genomic regions known to resist post-fertilization reprogramming. These include:
- Imprinted Control Regions (ICRs): Hypermethylation at paternally imprinted genes like H19, or hypomethylation at maternally imprinted genes like MEST and SNRPN, is strongly linked to impaired embryogenesis and diseases like Beckwith-Wiedemann syndrome [4] [26] [43].
- Transposable Elements: Regions like L1HS transposons often escape reprogramming [105] [26].
- Developmental Gene Promoters: Sperm histones are retained at promoters of genes critical for embryogenesis (e.g., marked by H3K4me3), and these can influence early gene expression in the embryo [37].
- Functional Validation: Use model systems to test the functional impact of observed epigenetic changes on gene expression and embryonic development [24].
Frequently Asked Questions (FAQs)
What are the key pillars of the sperm epigenome that should be reported?
A complete report should include data on three major epigenetic pillars [4] [26]:
- DNA Methylation: The presence of a methyl group on cytosine bases, primarily in CpG islands. It regulates genomic imprinting, transposon silencing, and gene expression [4] [43].
- Histone Modifications: Post-translational modifications (e.g., acetylation, methylation) on the retained ~1-15% of nucleosomes in mature sperm. These marks are enriched at gene promoters and enhancers important for development [37].
- Small Non-Coding RNAs (sncRNAs): A population of RNAs including miRNAs, tsRNAs, and rsRNAs. Their expression profiles in sperm can be altered by paternal environment and are implicated in intergenerational inheritance [26] [24].
Can lifestyle factors truly alter the sperm epigenome?
Yes, strong evidence shows that paternal preconception lifestyle and environment can dynamically reshape the sperm epigenome [105] [26]. These changes can affect sperm function and potentially influence offspring health.
Table: Paternal Environmental Exposures and Their Epigenetic Impacts
| Exposure | Documented Epigenetic Changes | Associated Offspring/Reproductive Outcomes |
|---|---|---|
| Obesity / High-Fat Diet [105] [26] | Altered DNA methylation and sncRNA profiles | Increased risk of metabolic dysfunction (impaired glucose tolerance, insulin resistance) in offspring [105] [26] |
| Psychological Stress [24] | Differential DNA Methylation Regions (DMRs); dysregulation of tsRNAs, miRNAs, and rsRNAs | Inherited behavioral, metabolic, and reproductive disorders in mouse models [24] |
| Smoking [26] | DNA hypermethylation in genes related to anti-oxidation and insulin resistance | Negative effects on sperm quality and offspring health [26] |
| Endocrine Disruptors [26] | Altered DNA methylation patterns during gametogenesis | Transgenerational transmission of infertility, obesity, and testicular disorders [26] |
| Exercise [105] | Altered DNA methylation near genes involved in nervous system development | Improved metabolic health; potential impact on offspring neurodevelopment [105] |
What is the difference between intergenerational and transgenerational inheritance?
It is critical to distinguish these terms in your reporting [24]:
- Intergenerational Inheritance: The transmission of a phenotypic or epigenetic trait from the directly exposed generation (F0) to their immediate offspring (F1). For paternal lineages, this is F0 to F1. The F1 germline was directly exposed in utero to the paternal environmental signal via the sperm [24].
- Transgenerational Inheritance: The transmission of a trait to generations that were never directly exposed to the original trigger. For paternal lineages, this is F2 and beyond. The F2 generation is the first to be derived from a germline that was not directly exposed, providing stronger evidence for true epigenetic inheritance [24].
Essential Experimental Protocols
This workflow is critical for obtaining pure sperm samples for epigenetic analysis.
Genome-Wide DNA Methylation Analysis Workflow
This general workflow outlines key steps for bisulfite sequencing, a gold-standard method for assessing DNA methylation.
Table: Essential Reagents for Sperm Epigenetic Studies
| Research Reagent | Function/Application | Key Considerations |
|---|---|---|
| Somatic Cell Lysis Buffer (SCLB) [30] | Selective lysis of contaminating leukocytes and somatic cells in semen. | Critical for sample purity. Composition: 0.1% SDS, 0.5% Triton X-100. Always prepare fresh. |
| Antibodies for Histone Modifications [37] | Chromatin Immunoprecipitation (ChIP) to map histone retention (e.g., H3K4me3, H3K27ac). | Must be validated for use in sperm; be aware of testis-specific histone variants. |
| Bisulfite Conversion Kit | Treats DNA to convert unmethylated cytosines to uracils, allowing methylation status to be read by sequencing or PCR. | Key step for DNA methylation analysis. Optimize conversion efficiency to avoid bias. |
| Protamine Removal Agents | Agents to decondense sperm chromatin for improved access to DNA and histones. | Required for most downstream applications. Over-digestion can damage the sample. |
| sncRNA Isolation Kits | Specialized kits for isolating and purifying small RNA fractions (tsRNAs, miRNAs, rsRNAs) from sperm. | Standard RNA kits may not efficiently recover the full spectrum of sncRNAs. |
Data Reporting Checklist
Adhere to this checklist to ensure comprehensive and reproducible reporting of sperm epigenetic data.
Sample Information & Quality Control
- Report participant demographics (age, BMI) and fertility status.
- Detail semen parameters (count, motility, morphology).
- Explicitly state the protocol used for somatic cell removal and provide metrics of purity (e.g., % somatic cells pre/post lysis).
- Report DNA/RNA integrity numbers (e.g., RIN, DIN).
Experimental Methods
- Specify the exact assay (e.g., Whole-Genome Bisulfite Sequencing, ChIP-seq, sncRNA-seq).
- List catalog numbers and batches for key reagents and antibodies.
- For sequencing, provide library preparation details and sequencing depth.
Data Analysis & Validation
- State the bioinformatic pipelines and software versions used.
- Define thresholds for calling differentially methylated regions (DMRs) or differentially expressed sncRNAs.
- For DMRs, report genomic coordinates (e.g., Hg38), gene associations, and mean methylation differences.
- Include validation data from an independent method (e.g., pyrosequencing for DNA methylation, qPCR for sncRNAs).
Conclusion
The standardization of sperm epigenetic protocols is not merely a technical necessity but a fundamental prerequisite for unlocking the full clinical potential of this field. By integrating foundational knowledge with robust methodologies, rigorous troubleshooting, and thorough validation, we can transform sperm epigenetics from a research tool into a reliable component of clinical diagnostics. This harmonized approach will enable accurate risk assessment, improve prognostic power for ART outcomes, and open new avenues for therapeutic interventions. Future efforts must focus on large-scale, multi-center collaborative studies to establish universal reference materials and data reporting standards, ultimately ensuring that insights into the paternal epigenetic legacy can be consistently and reliably applied to improve human health across generations.
References
- 1. Epigenetic regulation by DNA methylation, histone ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12500515/]
- 2. Epigenetics and male reproduction: the consequences of ... [https://clinicalepigeneticsjournal.biomedcentral.com/articles/10.1186/s13148-015-0155-4]
- 3. Sperm-borne small non-coding RNAs: potential functions and ... [https://cellandbioscience.biomedcentral.com/articles/10.1186/s13578-025-01347-4]
- 4. Sperm epigenetics and male infertility - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC11149391/]
- 5. Evaluating the challenges and reproducibility of studies ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8978984/]
- 6. Sperm DNA Methylation Epimutation Biomarkers for Male ... [https://www.nature.com/articles/s41598-019-52903-1]
- 7. Sperm is epigenetically programmed to regulate gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4971762/]
- 8. Exploring the Epigenetic Landscape of Spermatozoa [https://www.mdpi.com/2076-3921/13/12/1520]
- 9. Chromatin state maps: new technologies, new insights - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2486450/]
- 10. DNA Methylation Analysis Support—Troubleshooting [https://www.thermofisher.com/us/en/home/technical-resources/technical-reference-library/epigenetics-support-center/dna-methylation-analysis-support/dna-methylation-analysis-support-troubleshooting.html]
- 11. H3K27me3-mediated epigenetic regulation of TET1 in the ... [https://www.nature.com/articles/s41598-025-13618-8]
- 12. Mechanisms of oxidative stress-induced sperm dysfunction [https://www.frontiersin.org/journals/endocrinology/articles/10.3389/fendo.2025.1520835/full]
- 13. Oxidative-Stress-Mediated Epigenetic Dysregulation in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11765119/]
- 14. Unravelling the epigenetic impact: Oxidative stress and its ... [https://www.sciencedirect.com/science/article/abs/pii/S0890623823002058]
- 15. Crosstalk Between Oxidative Stress and Epigenetics [https://www.mdpi.com/2073-4409/13/22/1846]
- 16. Adaptation and changing phenotypes through ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12351697/]
- 17. The Role of Oxidative Stress in Epigenetic Changes ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8982122/]
- 18. Sperm sequencing reveals extensive positive selection in ... [https://www.nature.com/articles/s41586-025-09448-3]
- 19. Harmful DNA changes in the sperm found to rise sharply ... [https://www.news-medical.net/news/20251008/Harmful-DNA-changes-in-the-sperm-found-to-rise-sharply-with-age.aspx]
- 20. Epigenetic Mechanisms of Paternal Stress in Offspring ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7837765/]
- 21. Developmental origins of health and disease: Impact ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10262906/]
- 22. Epigenetic effects of paternal environmental exposures ... [https://www.sciencedirect.com/science/article/abs/pii/S0168952525001076]
- 23. Transgenerational Epigenetic Inheritance: myths and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4020004/]
- 24. Sperm epigenetic alterations contribute to inter [https://www.nature.com/articles/s41421-021-00343-5]
- 25. Pre-conceptional paternal diet impacts on offspring ... [https://www.nature.com/articles/s44324-024-00011-8]
- 26. How do lifestyle and environmental factors influence the ... [https://clinicalepigeneticsjournal.biomedcentral.com/articles/10.1186/s13148-025-01815-1]
- 27. How a Dating Podcast Made Me Question Sperm's Epigenetic ... [https://davissciencesays.ucdavis.edu/blog/how-dating-podcast-made-me-question-sperms-epigenetic-integrity]
- 28. Increasing age in men is negatively associated with sperm ... [https://www.frontiersin.org/journals/aging/articles/10.3389/fragi.2025.1603916/full]
- 29. THE GHOST IN OUR GENES: LEGAL AND ETHICAL ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3034450/]
- 30. Tackling somatic DNA contamination in sperm epigenetic ... [https://www.frontiersin.org/journals/reproductive-health/articles/10.3389/frph.2025.1506117/full]
- 31. Comprehensive multi-omics analysis uncovers potential risks ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12374335/]
- 32. Sperm DNA integrity testing: what is it? patient education ... [https://www.reproductivefacts.org/news-and-publications/fact-sheets-and-infographics/sperm-dna-integrity-testing-what-is-it/]
- 33. What should be done for men with sperm DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6125150/]
- 34. At-Home Semen Collection Instructions - Houston [https://www.inovifertility.com/blog/at-home-semen-collection-instructions-houston/]
- 35. Comprehensive guidance for human embryology, ... [https://www.asrm.org/practice-guidance/practice-committee-documents/comprehensive-guidance-for-human-embryology-andrology-and-endocrinology-laboratories-management-and-operations-a-committee-opinion-2022/]
- 36. Sperm Cryopreservation Today: Approaches, Efficiency, ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10296824/]
- 37. Emerging evidence that the mammalian sperm epigenome ... [https://www.nature.com/articles/s41467-023-37820-2]
- 38. A Comprehensive Guide to DNA Methylation Sequencing ... [https://www.epicypher.com/resources/blog/a-comprehensive-guide-to-dna-methylation-sequencing-methods/]
- 39. Comparison of current methods for genome-wide DNA ... [https://epigeneticsandchromatin.biomedcentral.com/articles/10.1186/s13072-025-00616-3]
- 40. Future of Methylation Arrays: Innovations and Applications [https://www.cd-genomics.com/resource-methylation-arrays-future-innovations-applications.html]
- 41. Comparison of current methods for genome-wide DNA ... [https://pubmed.ncbi.nlm.nih.gov/40855329/]
- 42. Comparative analysis of genome-scale, base-resolution ... [https://www.nature.com/articles/s41467-022-34828-y]
- 43. Sperm epigenetics and male infertility - Human Genomics [https://humgenomics.biomedcentral.com/articles/10.1186/s40246-024-00626-4]
- 44. Exploring the expanding universe of small RNAs - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9035129/]
- 45. A dataset of hidden small non-coding RNA in the testis ... [https://www.nature.com/articles/s41597-024-03612-6]
- 46. A novel diagnostic signature of circulating tsRNAs and ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267023007419]
- 47. Comprehensive profiling of tsRNAs in acute coronary ... [https://link.springer.com/article/10.1007/s00439-025-02742-0]
- 48. Comparative performance evaluation of bisulfite [https://pmc.ncbi.nlm.nih.gov/articles/PMC11969950/]
- 49. Comprehensive guide for epigenetics and transcriptomics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11799959/]
- 50. Conversion efficiency - Bismark [https://felixkrueger.github.io/Bismark/faq/conversion_efficiency/]
- 51. EM-seq Troubleshooting Guide [https://www.neb.com/en-us/tools-and-resources/troubleshooting-guides/em-seq-troubleshooting-guide?srsltid=AfmBOoorMm5pgih7ybnhq4dmsVebyemuxDJbB4V39Di9exXMWtkXttVO]
- 52. BCREval: a computational method to estimate the bisulfite ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-3334-z]
- 53. Bisulfite Conversion of DNA - Research journals - PLOS [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0135058]
- 54. Designing and interpreting 'multi-omic' experiments that may ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7477987/]
- 55. Multi-omics data integration for the identification of ... [https://pubmed.ncbi.nlm.nih.gov/38394258/]
- 56. Comprehensive multi-omics analysis uncovers potential risks ... [https://bmcbiol.biomedcentral.com/articles/10.1186/s12915-025-02379-5]
- 57. Sperm epigenomics: challenges and opportunities - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4166955/]
- 58. Ten quick tips for avoiding pitfalls in multi-omics data ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1011224]
- 59. Present and future challenges for the investigation of ... [https://www.sciencedirect.com/science/article/pii/S0160412023000491]
- 60. Preanalytical Errors in Clinical Laboratory Testing at a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10981510/]
- 61. Pre-Analytical Variation | myadlm.org [https://myadlm.org/cln/articles/2016/july/preanalytical-variation-the-leading-cause-of-error-in-laboratory-medicine]
- 62. One day is better than four days of ejaculatory abstinence ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8812405/]
- 63. Epigenetic age prediction in semen – marker selection and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8386575/]
- 64. Epigenetic determinants of reproductive potential augment ... [https://www.sciencedirect.com/science/article/pii/S2666335X23000496]
- 65. Collecting a Semen Sample [https://progyny.com/education/male-infertility/collecting-a-semen-sample/]
- 66. The role of epigenetics in spermatogenesis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4548616/]
- 67. Tackling somatic DNA contamination in sperm epigenetic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11835817/]
- 68. Design and validation of novel flow cytometry panels to ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2024.1432816/full]
- 69. A Paired Flow Cytometry–Pathology Assessment for Immune ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12566358/]
- 70. optimized identification and characterization of small RNAs [https://www.rna-seqblog.com/pandora-seq-optimized-identification-and-characterization-of-small-rnas/]
- 71. a bioinformatic pipeline for the analysis of small chimeric RNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9808574/]
- 72. A novel approach toward optimal workflow selection for DNA ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-024-05658-0]
- 73. Bioinformatics Pipeline Troubleshooting [https://www.meegle.com/en_us/topics/bioinformatics-pipeline/bioinformatics-pipeline-troubleshooting]
- 74. Troubleshooting Pipelines Guide - GitHub Pages [https://phac-nml.github.io/irida-documentation/user/administrator/troubleshooting/pipelines/]
- 75. A survey of the approaches for identifying differential ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6171488/]
- 76. An incremental framework for batch effect correction in DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12495439/]
- 77. Assessing and mitigating batch effects in large-scale omics ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03401-9]
- 78. Correcting batch effects in large-scale multiomics studies ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10483871/]
- 79. Batch Effect in Single-cell RNA-seq: Frequently Asked ... [https://www.elucidata.io/blog/batch-effect-in-single-cell-rna-seq-frequently-asked-questions-and-answers]
- 80. ComBat-met: adjusting batch effects in DNA methylation data [https://pmc.ncbi.nlm.nih.gov/articles/PMC12086544/]
- 81. Integration of multi-omics profiling reveals an epigenetic ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1540477/full]
- 82. How to Troubleshoot Sequencing Preparation Errors (NGS ... [https://www.cd-genomics.com/resource-sequencing-preparation-troubleshooting.html]
- 83. DNA methylation biomarkers to identify epigenetically ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10498810/]
- 84. Bringing epigenetics into the diagnostics of the andrology ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4215682/]
- 85. Improved quality metrics for association and reproducibility in ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05553-0]
- 86. Epigenetic homogeneity in histone methylation underlies ... [https://www.nature.com/articles/s41467-020-17238-w]
- 87. Epigenetic determinants of reproductive potential augment ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10843460/]
- 88. Epigenetics of Male Infertility: The Role of DNA Methylation [https://pmc.ncbi.nlm.nih.gov/articles/PMC8339558/]
- 89. Clinical promise and applications of epigenetic biomarkers [https://pmc.ncbi.nlm.nih.gov/articles/PMC11682640/]
- 90. testing an epigenetic clock to forecast in vitro fertilization ... [https://rbej.biomedcentral.com/articles/10.1186/s12958-025-01429-5]
- 91. Verification, analytical validation, and clinical validation (V3) [https://www.nature.com/articles/s41746-020-0260-4]
- 92. Reframing analytical validation in preimplantation genetic ... [https://pubmed.ncbi.nlm.nih.gov/41106951/]
- 93. An Expert Review From the French BLEFCO Group [https://pubmed.ncbi.nlm.nih.gov/41185394/]
- 94. Award-Winning Science: Path Fertility Recognized at ... [https://www.inherentbio.com/news/asrm-2025]
- 95. DNA methylation and machine learning - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC12512482/]
- 96. Harbinger Health Unveils Promising New Data on Early ... [https://harbinger-health.com/2025/04/25/harbinger-health-unveils-promising-new-data-on-early-cancer-detection-platform-at-aacr-2025/?utm_content=333731664&utm_medium=social&utm_source=linkedin&hss_channel=lcp-77681026]
- 97. EnsembleAge: enhancing epigenetic age assessment with ... [https://link.springer.com/article/10.1007/s11357-025-01808-1]
- 98. Chromatin Immunoprecipitation Troubleshooting Guide [https://www.cellsignal.com/learn-and-support/troubleshooting/chip-troubleshooting-guide?srsltid=AfmBOoqCS7NlhFi5WHBDXEutH9cuxZuKAFkelFNyMAciCA2IB4TOqT7t]
- 99. Mapping epigenetic modifications by sequencing ... [https://www.nature.com/articles/s41418-023-01213-1]
- 100. Experimental Approaches to the Study of Epigenomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2838972/]
- 101. The Combined Expression Profiles of Epigenetic ... [https://www.mdpi.com/2073-4425/16/11/1314]
- 102. Review reveals how paternal lifestyle shapes sperm ... [https://www.news-medical.net/news/20251017/Review-reveals-how-paternal-lifestyle-shapes-sperm-epigenetics-and-offspring-health.aspx]
- 103. Predicting male fertility from the sperm methylome [https://clinicalepigeneticsjournal.biomedcentral.com/articles/10.1186/s13148-022-01275-x]
- 104. Sperm chromatin: Evaluation, epigenetic signatures and ... [https://www.sciencedirect.com/science/article/pii/S0171933524000463]
- 105. Sperm epigenetics and influence of environmental factors - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6034033/]