Viral vs. Non-Viral Vectors for Gene Therapy: A Comprehensive Guide for Drug Development
This article provides a detailed comparison of viral and non-viral somatic gene therapy (SMGT) methods for researchers and drug development professionals.
Viral vs. Non-Viral Vectors for Gene Therapy: A Comprehensive Guide for Drug Development
Abstract
This article provides a detailed comparison of viral and non-viral somatic gene therapy (SMGT) methods for researchers and drug development professionals. It covers the foundational biology of leading vector platforms, their methodological applications in clinical settings, strategies for troubleshooting critical challenges like immunogenicity and manufacturing, and a direct validation of their relative strengths and weaknesses. Synthesizing the latest clinical data and technological advances, the review offers a strategic framework for selecting and optimizing gene delivery systems to accelerate therapeutic development from the lab to the clinic.
Understanding Gene Delivery Vectors: Core Principles and System Classifications
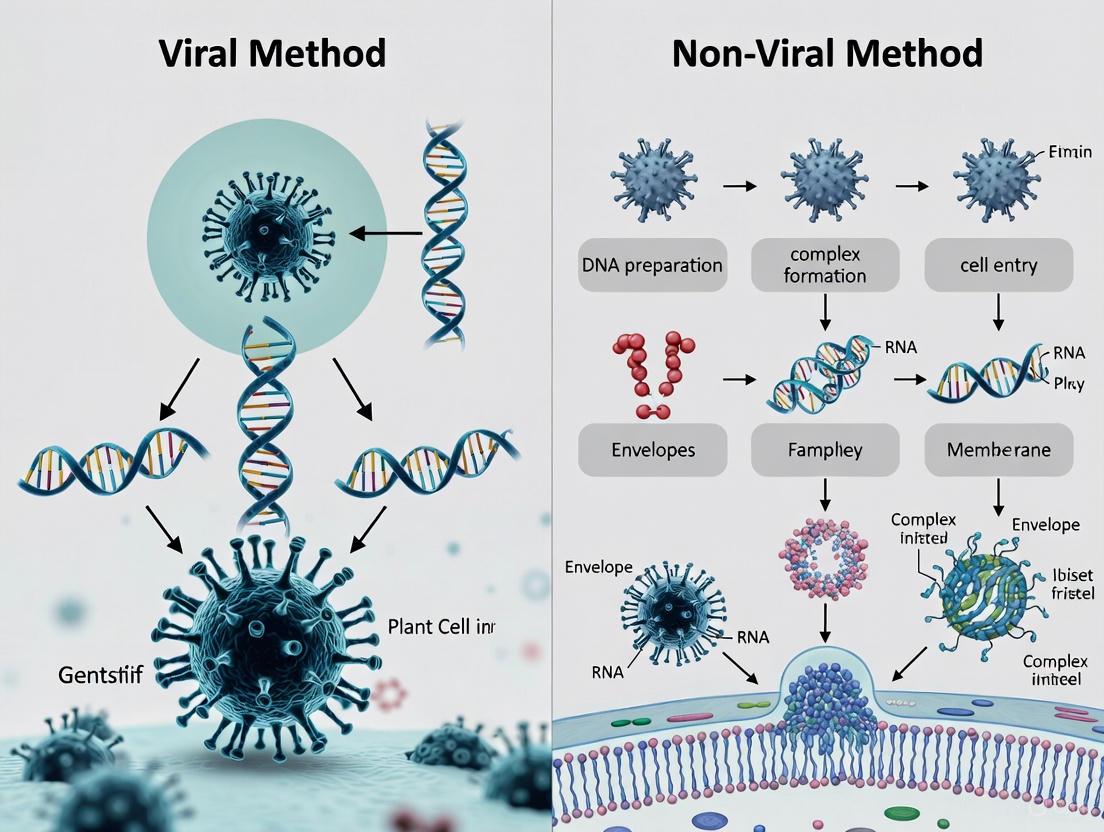
The remarkable progress in somatic gene therapy over the past decades has fundamentally shifted treatment paradigms for numerous genetic disorders, offering potential cures for conditions previously considered untreatable. At the heart of this therapeutic revolution are the delivery vectors—sophisticated vehicles designed to transport therapeutic genetic material into target cells. These vectors broadly fall into two categories: viral vectors, which harness the natural efficiency of viruses, and non-viral vectors, which employ synthetic or biological compounds for gene delivery [1] [2]. The choice between viral and non-viral delivery systems represents a critical decision point in therapeutic development, balancing factors including delivery efficiency, cargo capacity, immunogenicity, and manufacturing scalability.
The field has witnessed significant milestones, with 35 vector-based therapies currently approved globally. Viral vectors dominate the approved therapies landscape, constituting 29 of these treatments, while non-viral approaches account for the remaining 6 approved products [1] [3]. This distribution reflects the current state of vector technology while highlighting the growing importance of non-viral platforms. This guide provides a comprehensive, objective comparison of these delivery systems, focusing on their performance characteristics, experimental applications, and practical implementation for researchers and drug development professionals.
Comparative Analysis of Vector Platforms
Viral Vector Systems: Established Workhorses of Gene Therapy
Viral vectors leverage the natural ability of viruses to efficiently deliver genetic material into cells. The three predominant viral platforms—lentivirus (LV), adenovirus (Ad), and adeno-associated virus (AAV)—each offer distinct advantages and limitations that make them suitable for different therapeutic applications [1] [3].
Lentiviral (LV) vectors, derived from retroviruses, are primarily utilized in ex vivo gene therapy applications, particularly in chimeric antigen receptor (CAR) T-cell therapies and hematopoietic stem cell gene therapies. Their key advantage is stable genomic integration, enabling long-term transgene expression in dividing cells. Approved LV-based therapies include Kymriah for leukemia, Zynteglo for β-thalassemia, and Libmeldy for metachromatic leukodystrophy [1] [3]. However, this integration capacity carries the risk of insertional mutagenesis, as evidenced by rare cases of blood cancer development following treatment with Skysona [3].
Adenoviral (Ad) vectors feature a large double-stranded DNA genome capable of accommodating payloads up to 36 kb, making them suitable for delivering large genetic sequences. They provide high transduction efficiency and robust transient transgene expression without genomic integration. Their primary limitation is significant immunogenicity, which can trigger potent inflammatory responses and has largely confined their clinical use to oncology and vaccine applications [3] [2]. Early successes include Gendicine and Oncorine for head and neck cancers [1].
Adeno-associated viral (AAV) vectors have emerged as the leading platform for in vivo gene therapy due to their favorable safety profile, including non-pathogenicity and primarily episomal persistence that reduces genotoxicity risks. Their ability to transduce non-dividing cells and provide long-term transgene expression has led to multiple approved therapies, including Luxturna for inherited retinal dystrophy, Zolgensma for spinal muscular atrophy, and Hemgenix for hemophilia B [1] [4] [3]. A significant limitation is their constrained cargo capacity (~4.7 kb), though innovative dual-vector approaches are being developed to deliver larger genes [3].
Non-Viral Vector Systems: Emerging Safe Alternatives
Non-viral vectors have gained substantial momentum as safer, more scalable alternatives to viral platforms, particularly following the successful deployment of lipid nanoparticles (LNPs) in mRNA COVID-19 vaccines [3] [5].
Lipid Nanoparticles (LNPs) represent the most advanced non-viral platform, comprising ionizable lipids, phospholipids, cholesterol, and PEG-lipids that self-assemble into vesicles capable of encapsulating and protecting nucleic acids. LNPs efficiently deliver their payload through endocytic pathways and have enabled approved therapies like Patisiran for hereditary transthyretin amyloidosis [1] [3]. They are also being investigated for CRISPR-Cas9 delivery, as demonstrated by NTLA-2002 for hereditary angioedema [3]. Key advantages include large cargo capacity, low immunogenicity, and scalable manufacturing, though challenges remain with lower transfection efficiency in certain tissues and predominant liver-focused biodistribution [3] [5].
N-Acetylgalactosamine (GalNAc) conjugates represent a specialized approach for liver-targeted delivery of RNA therapeutics. GalNAc, a ligand for the asialoglycoprotein receptor highly expressed on hepatocytes, enables efficient receptor-mediated uptake of conjugated RNA molecules. This platform has enabled multiple FDA-approved drugs, including Givlaari for acute hepatic porphyria, Oxlumo for primary hyperoxaluria type 1, and Leqvio for hypercholesterolemia [1] [3]. The primary advantage is tissue-specific targeting with subcutaneous administration, though application is currently restricted to hepatic conditions [3].
Table 1: Comparative Analysis of Major Gene Therapy Vector Platforms
| Vector Platform | Cargo Capacity | Integration Profile | Immunogenicity | Primary Applications | Key Advantages | Major Limitations |
|---|---|---|---|---|---|---|
| Lentivirus (LV) | ~8 kb | Stable integration | Moderate | Ex vivo therapies (CAR-T, hematopoietic stem cells) | Long-term expression in dividing cells | Risk of insertional mutagenesis |
| Adenovirus (Ad) | Up to 36 kb | Episomal | High | Cancer therapy, vaccines | Large cargo capacity, high transduction efficiency | Strong immune response |
| AAV | ~4.7 kb | Predominantly episomal | Low to moderate | In vivo gene therapy (neuromuscular, ocular, hepatic) | Excellent safety profile, transduces non-dividing cells | Limited cargo capacity, pre-existing immunity |
| LNP | >10 kb | Non-integrating | Low | siRNA, mRNA, CRISPR delivery | Large cargo capacity, scalable manufacturing | Mostly liver tropism, lower efficiency in some tissues |
| GalNAc | Variable | Non-integrating | Very low | Liver-targeted RNA therapeutics | High tissue specificity, subcutaneous administration | Restricted to hepatic applications |
Experimental Data and Performance Metrics
Quantitative Comparison of Delivery Efficiency
Rigorous evaluation of vector performance requires standardized metrics across multiple parameters. The following table synthesizes experimental data from preclinical and clinical studies to enable direct comparison of key performance indicators.
Table 2: Performance Metrics of Vector Platforms in Preclinical and Clinical Applications
| Vector Platform | Transduction Efficiency In Vivo | Expression Duration | Therapeutic Dose Range | Clinical Success Rate* | Manufacturing Titer |
|---|---|---|---|---|---|
| LV (ex vivo) | >80% (in modified cells) | Long-term (years) | 1-10×10^6 transduced cells/kg | High (>70% in approved therapies) | 10^8-10^9 TU/mL |
| Ad | 60-95% (varies by tissue) | Transient (weeks) | 10^11-10^13 VP | Moderate in oncology | 10^11-10^12 VP/mL |
| AAV | 20-90% (serotype-dependent) | Long-term (years) | 10^11-10^15 VP (route-dependent) | High (>80% in approved therapies) | 10^13-10^14 VG/L |
| LNP | 15-60% (liver) | Transient (days-weeks) | 0.1-1.0 mg/kg mRNA | High in approved indications | Highly scalable |
| GalNAc | 40-80% (hepatocytes) | Transient (weeks-months) | 1-10 mg/kg (subcutaneous) | High in approved indications | Highly scalable |
*Clinical success rate defined as percentage of patients achieving primary efficacy endpoint in registrational trials for approved products.
Key Experimental Findings by Vector Class
Viral Vector Performance: AAV vectors demonstrate particularly impressive long-term transduction in clinical applications, with sustained transgene expression documented for over 5 years following single administration in retinal and neuromuscular disorders [4]. However, dose-dependent immune responses remain a significant concern, particularly with high-dose systemic administration [6]. Recent clinical data reveal that both innate and adaptive immune responses to AAV capsids can substantially impact therapeutic efficacy, with pre-existing neutralizing antibodies excluding approximately 30-50% of potential patients from systemic AAV therapy trials [6].
LV vectors exhibit remarkable ex vivo transduction efficiency, with studies demonstrating >80% CAR integration in T-cells across multiple clinical trials [1]. The recent development of LV-based therapies for sickle cell disease and β-thalassemia has demonstrated sustained hemoglobin production years after treatment, highlighting their potential for durable effect [1] [3].
Non-Viral Vector Performance: LNP delivery systems have shown dose-dependent transfection efficiency in preclinical models, with Tessera Therapeutics reporting approximately 42% CAR integration in ex vivo T-cell editing studies using proprietary LNPs [5]. Notably, their technology achieved multiplexed knock-out at TRAC and B2M loci with >88% efficiency while simultaneously enabling CAR integration at 20% efficiency [5].
GalNAc-conjugated therapies demonstrate exceptional hepatocyte specificity, with clinical studies showing >80% target gene knockdown in the liver at doses as low as 1-10 mg/kg administered subcutaneously [3]. This approach minimizes off-target effects while maintaining potent on-target activity.
Experimental Protocols and Methodologies
Standardized Assessment of Vector Performance
To ensure reproducible evaluation across vector platforms, researchers should implement standardized experimental protocols for key performance parameters.
Protocol 1: In Vivo Transduction Efficiency Assessment
- Objective: Quantify vector-mediated gene delivery efficiency in target tissues
- Procedure:
- Administer vectors via relevant route (IV, IT, IM, etc.) at standardized doses
- Harvest target tissues at predetermined endpoints (e.g., 7, 14, 28 days post-administration)
- Process tissues for:
- qPCR/RT-qPCR: Quantify vector genome copies and transgene expression
- Immunohistochemistry: Localize and quantify transgene expression
- Western Blot: Confirm functional protein production
- Calculate transduction efficiency as percentage of target cells expressing transgene
Protocol 2: Immune Response Profiling
- Objective: Characterize innate and adaptive immune responses to vector administration
- Procedure:
- Collect serial blood samples pre- and post-vector administration
- Analyze samples for:
- Cytokine profiling (IFN-γ, IL-6, TNF-α) via ELISA or multiplex assays
- Neutralizing antibody detection using in vitro transduction inhibition assays
- T-cell responses via ELISpot for IFN-γ secretion upon capsid stimulation
- Perform immunohistochemistry on tissues for immune cell infiltration assessment
Protocol 3: Biodistribution and Persistence Studies
- Objective: Track vector distribution and transgene persistence over time
- Procedure:
- Administer radiolabeled or barcoded vectors
- Quantify vector genomes in target and non-target tissues at multiple timepoints
- Assess transgene expression duration via longitudinal imaging (if reporter included) or terminal tissue analysis
- For integrating vectors, analyze integration sites using LAM-PCR or next-generation sequencing methods
Specialized Methodologies by Vector Class
Viral Vector-Specific Protocols:
- Empty Capsid Contamination Assessment: Critical for AAV preparations, performed via analytical ultracentrifugation or ELISA to quantify ratio of full to empty particles, which impacts potency and immunogenicity [6].
- Replication-Competent Virus Testing: Essential safety assessment for all viral vector lots via co-culture assays with permissive cell lines and PCR detection.
- Serotype-Specific Tropism Profiling: Comparative evaluation of different AAV serotypes (AAV1, AAV2, AAV8, AAV9, etc.) for tissue-specific transduction efficiency using barcoded libraries [4] [7].
Non-Viral Vector-Specific Protocols:
- LNP Characterization: Comprehensive physical assessment including particle size (dynamic light scattering), encapsulation efficiency (Ribogreen assay), polydispersity index, and zeta potential.
- Endosomal Escape Efficiency: Quantification of functional delivery using split-luciferase or fluorophore-based systems that require cytosolic delivery for signal generation.
- Tropism Modification Assessment: Evaluation of targeting ligand incorporation through comparative biodistribution studies in relevant animal models.
Visualization of Key Mechanisms and Workflows
Viral Vector Mechanism and Immune Recognition
The following diagram illustrates the intracellular trafficking mechanisms of viral vectors and key immune recognition pathways that impact therapeutic efficacy.
Diagram 1: Viral vector mechanisms and immune recognition. This diagram illustrates the intracellular trafficking of viral vectors (top) and the immune recognition pathways (bottom) that can limit therapeutic efficacy, including TLR9-mediated recognition of CpG motifs and subsequent neutralizing antibody production.
Non-Viral Vector Delivery Workflow
The following diagram outlines the complete workflow for non-viral vector delivery, from formulation to therapeutic action.
Diagram 2: Non-viral vector delivery workflow. This diagram illustrates the complete pathway for non-viral vector delivery, including LNP formulation (left), cellular delivery and processing (center), and therapeutic action mechanisms (right), with GalNAc-targeted delivery as a specialized approach.
The Scientist's Toolkit: Essential Research Reagents
Successful gene therapy research requires carefully selected reagents and materials optimized for specific vector platforms. The following table details essential research tools for investigating viral and non-viral delivery systems.
Table 3: Essential Research Reagents for Gene Therapy Vector Research
| Reagent/Material | Function | Key Considerations | Example Applications |
|---|---|---|---|
| Packaging Plasmids | Essential components for recombinant viral vector production | Optimize ratio for maximum titer and full capsid percentage | AAV, LV, Ad vector production |
| Cell Lines for Production | Factory cells for viral vector generation | Select for permissiveness, scalability, and low adventitious agents | HEK293, Sf9 systems for AAV production |
| Purification Systems | Isolation and concentration of vectors from crude lysates | Balance purity, yield, and activity preservation | AAV purification using affinity or ion-exchange chromatography |
| Characterization Assays | Quality control and vector quantification | Standardize across batches for reproducibility | qPCR for genome titer, ELISA for capsid titer, TCID50 for infectivity |
| Ionizable Lipids | Core component of LNP formulations for nucleic acid encapsulation | Structure affects efficacy, biodegradability, and toxicity | SM-102, DLin-MC3-DMA for mRNA delivery |
| Targeting Ligands | Enable cell-specific delivery of non-viral vectors | Conjugation method impacts activity and stability | GalNAc for hepatocytes, peptide ligands for extrahepatic targeting |
| Animal Models | In vivo evaluation of biodistribution and efficacy | Consider species-specific tropism and immune responses | Mice, NHP for AAV studies; disease-specific models for efficacy |
| Akt-IN-18 | Akt-IN-18, MF:C19H14ClN5O2S, MW:411.9 g/mol | Chemical Reagent | Bench Chemicals |
| Mao-B-IN-31 | Mao-B-IN-31, MF:C16H14N2O2S, MW:298.4 g/mol | Chemical Reagent | Bench Chemicals |
The evolving landscape of gene therapy vectors reflects a dynamic interplay between viral and non-viral platforms, each advancing to address persistent challenges. Viral vectors continue to dominate clinical applications, with ongoing innovations focused on capsid engineering to evade pre-existing immunity, tropism refinement for enhanced tissue specificity, and dual-vector systems to overcome cargo limitations [1] [6]. Non-viral platforms are rapidly evolving with novel ionizable lipids improving delivery efficiency beyond the liver, biodegradable formulations enhancing safety profiles, and modular design approaches enabling precise targeting of extrahepatic tissues [3] [5].
The future of somatic gene therapy delivery will likely witness increased convergence between viral and non-viral technologies, incorporating viral elements into synthetic vectors and applying non-viral principles to improve viral vector manufacturing. As both platforms mature, the selection criteria will increasingly focus on matching vector capabilities to specific therapeutic contexts—considering disease pathophysiology, target tissue accessibility, required expression level and duration, and patient-specific factors such as pre-existing immunity. This nuanced, application-driven approach to vector selection will ultimately expand the therapeutic landscape, bringing transformative treatments to broader patient populations across diverse genetic disorders.
Viral vectors are engineered viruses designed to deliver therapeutic genetic material into human cells, serving as a cornerstone of modern gene therapy [8]. By harnessing the innate ability of viruses to infect cells and introduce their genetic payload, scientists have developed powerful tools to treat a wide range of genetic disorders, cancers, and infectious diseases [9]. Among the various options available, three viral vector platforms have emerged as predominant in both research and clinical applications: lentiviruses (LV), adenoviruses (Ad), and adeno-associated viruses (AAV) [10] [11]. Each vector system possesses distinct biological characteristics, performance parameters, and application landscapes that researchers must carefully consider when designing gene therapy strategies [11]. The selection of an appropriate viral vector is critical not only for experimental success but also for clinical efficacy and safety, as mismatches between vector capabilities and therapeutic requirements can lead to failed experiments or adverse patient outcomes [10]. This guide provides a comprehensive, data-driven comparison of these three viral vector systems to inform researchers, scientists, and drug development professionals in their experimental design and therapeutic development decisions.
Comparative Analysis of Key Vector Characteristics
The effective deployment of viral vectors in gene therapy requires a thorough understanding of their fundamental biological properties and performance capabilities. The table below summarizes the critical differentiating characteristics of AAV, lentiviral, and adenoviral vectors, enabling researchers to make informed decisions based on their specific experimental or therapeutic requirements.
Table 1: Key Characteristics of AAV, Lentivirus, and Adenovirus Vectors
| Characteristic | AAV | Lentivirus (LV) | Adenovirus (Ad) |
|---|---|---|---|
| Genomic Material | Single-stranded DNA (ssDNA) [12] | RNA [13] | Double-stranded DNA (dsDNA) [10] |
| Packaging Capacity | ~4.7 kb [10] [13] [12] | ~8 kb [10] [11] | Up to ~36 kb [10] |
| Genome Integration | Predominantly non-integrating (episomal) [13] [12] | Integrating [11] [13] | Non-integrating (episomal) [11] |
| Expression Kinetics | Slow onset, but long-term (months to years) [10] | Slow onset, stable long-term expression [10] | Rapid onset, transient expression (days to weeks) [10] [11] |
| Immunogenicity | Low [11] [13] | Low [10] | High [10] [11] |
| Tropism (Infection Range) | Broad, with serotype-specific targeting [12] | Broad (dividing and non-dividing cells) [11] [13] | Broad (dividing and non-dividing cells) [10] [9] |
| Primary Applications | In vivo gene therapy for monogenic diseases [13] [12] | Ex vivo cell engineering (e.g., CAR-T, HSCs) [10] [13] | Vaccines, oncolytic therapy, transient gene expression [10] [11] |
| Biosafety Level | BSL-1 [10] | BSL-2 [10] | BSL-2 [10] |
Critical Performance Differentiators
Packaging Capacity and Transgene Design: The limited ~4.7 kb capacity of AAV vectors presents significant constraints for delivering large genetic sequences, sometimes necessitating dual-vector approaches for larger genes [10] [12]. Lentiviral vectors offer a middle ground with approximately 8 kb capacity, while adenoviral vectors provide the most flexibility with the ability to accommodate large or multiple transgenes up to 36 kb [10] [11]. This capacity directly influences experimental design, particularly for complex expression cassettes requiring large regulatory elements or multiple transcriptional units.
Expression Duration and Stability: The persistence of transgene expression varies dramatically between vector systems, dictated by their fundamental biology. AAV vectors achieve long-term expression through episomal persistence in non-dividing cells, often lasting months to years [10]. Lentiviral vectors provide stable long-term expression through integration into the host genome, ensuring persistence through cell division [11]. Adenoviral vectors generate high-level but transient expression due to episomal maintenance and immune-mediated clearance of transduced cells, typically lasting days to weeks [10] [11].
Safety Considerations: AAV vectors demonstrate the most favorable safety profile with low immunogenicity and minimal risk of insertional mutagenesis due to their predominantly episomal persistence [10] [13]. Lentiviral vectors present a theoretical risk of insertional mutagenesis, though this is significantly lower than earlier retroviral vectors due to preferential integration into active transcriptional units [13]. Adenoviral vectors trigger robust immune responses that can eliminate transduced cells and complicate repeated administration, though this property can be advantageous in vaccine and oncolytic applications [10] [11].
Application-Specific Vector Selection Guide
The therapeutic or research objective represents the primary determinant in viral vector selection. Different disease targets, target tissues, and expression requirements necessitate careful matching with vector capabilities. The following table outlines optimal application domains for each vector system based on their inherent biological properties and documented clinical successes.
Table 2: Application-Based Vector Selection Guide
| Therapeutic/Research Area | Recommended Vector | Rationale and Evidence |
|---|---|---|
| Neurological Disorders (e.g., Parkinson's, Alzheimer's) | AAV [10] | Ability to cross the blood-brain barrier; long-term expression in non-dividing neurons; multiple CNS-tropic serotypes (AAV9, AAVrh.10) [10] [12]. |
| Ophthalmic Diseases (e.g., LCA2) | AAV [10] [12] | Successful clinical application (Luxturna); low immunogenicity in immune-privileged eye; sustained expression in retinal cells [14] [12]. |
| Hematological Disorders & ex vivo Cell Therapy (e.g., SCID, CAR-T) | Lentivirus [10] [13] | Stable integration in hematopoietic stem/progenitor cells ensures persistence in differentiated lineages; clinical success in β-thalassemia and CAR-T therapies [10] [13]. |
| Vaccine Development | Adenovirus [10] | Strong immunogenicity stimulates robust immune response; rapid, high-level antigen expression; successful platform for COVID-19 vaccines [10]. |
| Muscular Dystrophies | AAV [11] | Efficient transduction of muscle tissue; long-term expression in post-mitotic myofibers; clinical use of AAV9 in limb-girdle muscular dystrophy trials [11] [12]. |
| Oncolytic Virotherapy | Adenovirus [11] | Natural tropism for epithelial cells; cytotoxic effects in cancer cells; ability to engineer replication-competent variants for selective tumor lysis [11]. |
| Large Gene Delivery (>5 kb) | Adenovirus or Lentivirus [10] | Large packaging capacity of adenovirus (up to 36 kb) and lentivirus (~8 kb) accommodates larger genetic payloads [10] [11]. |
Tissue Tropism and Targeting Strategies
AAV Serotype Selection: Natural AAV serotypes display distinct tissue tropisms that can be leveraged for targeted delivery. For example, AAV2 demonstrates broad tropism, AAV8 and AAV9 show strong liver and muscle tropism, while AAV5 efficiently transduces photoreceptors and airway epithelial cells [12]. Engineered capsid variants are expanding these native tropisms to enable more precise targeting [13] [12].
Lentiviral Pseudotyping: The tropism of lentiviral vectors can be redirected through pseudotyping with envelope proteins from other viruses. The most common approach utilizes the Vesicular Stomatitis Virus G-protein (VSV-G), which confers broad tropism across many cell types [13]. Alternative envelopes from rabies virus, measles virus, or other families can redirect transduction to specific neuronal subsets or other specialized cell types.
Adenovirus Fiber Modifications: Natural adenovirus tropism is primarily determined by interactions between the viral fiber knob domain and cellular receptors (CAR, CD46). Genetic modification of the fiber protein enables retargeting to specific cell surface markers, enhancing specificity and reducing off-target transduction [10].
Experimental Design and Methodological Considerations
Vector Engineering and Production Workflows
The production of high-quality viral vectors requires specialized methodologies that differ significantly between systems. Below is a standardized workflow for AAV production, one of the most complex manufacturing processes, highlighting key stages where lentiviral and adenoviral production would diverge.
Diagram 1: Viral vector production workflow highlighting key stages and plasmid requirements for different systems. AAV production requires a 3-plasmid system (red), while lentiviral production typically uses a 4-plasmid system (green).
Key Experimental Protocols
AAV Vector Production via Transient Transfection
Principle: The most common AAV production method uses HEK293 cells that are transfected with three plasmids: (1) the transgene plasmid containing ITRs, (2) the Rep/Cap plasmid providing replication and capsid proteins, and (3) the adenoviral helper plasmid providing essential helper functions [12].
Detailed Methodology:
- Cell Culture: Maintain HEK293 cells in DMEM with 10% FBS at 37°C, 5% CO₂. Seed cells at 70-80% confluence in cell factories or multilayer flasks for large-scale production.
- Plasmid Transfection: Use polyethylenimine (PEI) or calcium phosphate precipitation to co-transfect the three plasmids at an optimized ratio (typically 1:1:1 mass ratio) when cells reach 80-90% confluence.
- Harvest: Collect cells 48-72 hours post-transfection by scraping and centrifugation at 500 × g for 10 minutes.
- Lysis: Resuspend cell pellet in lysis buffer (150 mM NaCl, 50 mM Tris-HCl, pH 8.5) and perform three freeze-thaw cycles between -80°C and 37°C.
- Benzonase Treatment: Incubate lysate with benzonase (50 U/mL) at 37°C for 30 minutes to digest unpackaged nucleic acids.
- Purification: Purify vectors using iodixanol gradient ultracentrifugation or affinity chromatography. For iodixanol gradients, centrifuge at 350,000 × g for 1 hour and collect the 40% iodixanol fraction containing viral particles.
- Concentration and Buffer Exchange: Use centrifugal concentrators for final concentration and exchange into formulation buffer (e.g., PBS with 0.001% Pluronic F-68).
- Quality Control: Determine vector genome titer by quantitative PCR, assess purity by SDS-PAGE, and test for adventitious agents.
Lentiviral Vector Transduction for Stable Cell Line Generation
Principle: Lentiviral vectors enable stable integration of transgenes through reverse transcription and integration into the host genome, making them ideal for creating stably modified cell lines [13].
Detailed Methodology:
- Vector Production: Produce lentiviral vectors by transfecting HEK293T cells with a 4-plasmid system (packaging, envelope, Rev, and transfer plasmids) using PEI transfection.
- Harvest and Concentration: Collect supernatant 48 and 72 hours post-transfection, filter through 0.45μm membrane, and concentrate by ultracentrifugation at 50,000 × g for 2 hours.
- Target Cell Preparation: Plate target cells at 30-50% confluence in growth medium supplemented with 8μg/mL polybrene to enhance transduction efficiency.
- Transduction: Add appropriate volume of lentiviral vector to achieve desired multiplicity of infection (MOI). Incubate for 24 hours.
- Selection and Expansion: Replace medium with selection antibiotic (e.g., puromycin, blasticidin) 48 hours post-transduction. Maintain selection pressure for 5-7 days until control cells are dead.
- Clonal Isolation: Isolate single cells by limiting dilution or FACS sorting into 96-well plates. Expand clones and screen for transgene expression.
- Validation: Validate stable integration by genomic PCR, Southern blot, or next-generation sequencing integration site analysis.
Adenoviral Vector Transduction for Transient High-Level Expression
Principle: Adenoviral vectors provide high transduction efficiency and rapid transgene expression, making them suitable for applications requiring high but transient protein production [10] [11].
Detailed Methodology:
- Cell Plating: Plate target cells at 70-80% confluence in appropriate growth medium 24 hours before transduction.
- Vector Thawing: Thaw adenoviral vector stock quickly at 37°C and place immediately on ice. Dilute in cold serum-free medium to desired concentration.
- Transduction: Remove growth medium from cells and add vector-containing medium. Incubate at 37°C for 1-2 hours with gentle rocking every 15 minutes.
- Medium Replacement: Replace transduction medium with complete growth medium and return cells to incubator.
- Expression Monitoring: Monitor transgene expression beginning 6-24 hours post-transduction, with peak expression typically occurring at 24-48 hours.
- Functional Assays: Perform downstream functional assays during peak expression window (24-72 hours).
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Viral Vector Applications
| Reagent/Category | Function | Example Applications |
|---|---|---|
| Packaging Plasmids | Provide essential viral genes in trans for vector production [12] | AAV Rep/Cap plasmids; LV Gag/Pol packaging plasmids [13] [12] |
| Helper Plasmids | Supply auxiliary viral functions needed for replication [12] | Adenoviral helper plasmids for AAV production [12] |
| Cell Lines | Specialized cells for vector production or transduction studies | HEK293/293T cells [12]; target cell lines (primary cells, stem cells) |
| Transfection Reagents | Facilitate plasmid DNA delivery into packaging cells | PEI, calcium phosphate, commercial lipid-based reagents [13] |
| Purification Matrices | Isolation and purification of viral vectors from crude lysates | Iodixanol gradients; affinity chromatography resins [13] |
| Titration Kits | Quantitative assessment of vector concentration | qPCR-based titer kits; physical particle detection assays |
| Transduction Enhancers | Improve vector uptake and gene transfer efficiency | Polybrene [13]; other cationic polymers |
| Tubulin inhibitor 33 | Tubulin inhibitor 33, MF:C24H22N4O3, MW:414.5 g/mol | Chemical Reagent |
| SARS-CoV-2-IN-74 | SARS-CoV-2-IN-74, MF:C26H33N3O3, MW:435.6 g/mol | Chemical Reagent |
Manufacturing, Safety, and Regulatory Perspectives
Production Scale-Up and Cost Considerations
The transition from research-scale to clinical-grade vector manufacturing presents significant challenges that vary considerably between vector platforms. The table below compares key manufacturing considerations that impact development timelines, production costs, and commercialization potential.
Table 4: Manufacturing and Commercialization Comparison
| Manufacturing Aspect | AAV | Lentivirus | Adenovirus |
|---|---|---|---|
| Production Complexity | High [10] | High [10] | Moderate [10] |
| Production Cost | High (raw materials account for 30-40% of cost) [10] | High [10] | Lower (50-60% less than AAV/LV) [10] |
| Scale-Up Challenges | Significant; requires strict control of cell culture conditions and purification parameters [10] | Significant; affected by cell density, transfection ratios, and requires high biosafety containment [10] | More straightforward; high replication enables large-scale production with standardized processes [10] |
| Common Impurities | Empty/partial capsids (significant productivity challenge) [13] | Replication-competent lentiviruses (RCL) [13] | Viral and host cell proteins, DNA [10] |
| Regulatory Precedents | Multiple approved products (e.g., Luxturna, Zolgensma) [13] [12] | Approved therapies (e.g., CAR-T products) [13] | Approved vaccines and oncolytic therapies [10] |
Safety Profiles and Risk Mitigation Strategies
AAV Vector Safety: AAV vectors present the most favorable safety profile with low immunogenicity and minimal risk of insertional mutagenesis due to predominantly episomal persistence [10] [13]. Primary safety concerns include dose-dependent immune responses, particularly at high systemic doses, and potential hepatotoxicity [12]. Preclinical studies include thorough biodistribution analysis and immune response characterization. Risk mitigation strategies include empty capsid removal, corticosteroid prophylaxis, and tissue-specific promoter use to limit off-target expression [12].
Lentiviral Vector Safety: While offering stable gene expression through genomic integration, lentiviral vectors present theoretical risks of insertional mutagenesis and oncogene activation [11] [13]. Modern self-inactivating (SIN) designs with deleted enhancer/promoter regions in the LTRs have significantly improved safety profiles [13]. Additional safety modifications include chromatin insulators to prevent transgene silencing and enhancer blocking. Regulatory requirements typically include integration site analysis in clinical trials to monitor for clonal dominance [10].
Adenoviral Vector Safety: The strong immunogenicity of adenoviral vectors represents both a therapeutic advantage (vaccines) and limitation (inflammatory toxicity) [10] [11]. First-generation vectors retaining viral genes elicit robust immune responses, while newer gutless designs with deleted viral coding sequences show improved safety profiles [10]. Preclinical evaluation includes thorough assessment of innate and adaptive immune responses, with dose escalation studies to establish therapeutic windows. Clinical monitoring focuses on inflammatory markers and organ function, particularly liver enzymes [10].
The selection of an optimal viral vector platform represents a critical decision point in gene therapy research and development, with significant implications for experimental success and therapeutic outcomes. AAV vectors excel in in vivo applications requiring long-term gene expression in non-dividing tissues with minimal immunogenicity, particularly for monogenic diseases affecting neurological, ocular, and muscular systems [10] [12]. Lentiviral vectors remain the gold standard for ex vivo cell engineering applications where stable genomic integration and persistent transgene expression through cell division are required, as evidenced by their success in CAR-T therapies and hematopoietic stem cell treatments [10] [13]. Adenoviral vectors provide unparalleled transduction efficiency and rapid, high-level transgene expression, making them ideal for vaccine development, oncolytic virotherapy, and applications requiring transient expression [10] [11].
Future directions in viral vector development focus on overcoming current limitations through capsid engineering to enhance tissue specificity and evade pre-existing immunity [13] [12], optimizing production processes to reduce costs and improve scalability [10] [13], and developing novel vector systems with enhanced safety profiles and larger cargo capacities. As the gene therapy field continues to evolve, the strategic selection and continued refinement of these viral vector platforms will remain essential for translating promising research into effective clinical therapies.
Gene therapy represents a paradigm shift in treating human diseases by addressing genetic defects at their root cause. The success of this intervention critically depends on the vectors that deliver therapeutic genetic material. While viral vectors have been the historical workhorse, accounting for 29 of the 35 approved vector-based therapies globally, non-viral vectors are gaining substantial momentum as safer, more scalable alternatives [1] [3]. These synthetic systems do not integrate into the host genome and typically elicit fewer immunogenic reactions than their viral counterparts [15]. Their broader cargo capacity and lower production costs make them particularly attractive for a wide range of therapeutic applications, from silencing disease-causing genes to encoding therapeutic proteins [3].
This guide focuses on three leading non-viral platforms: Lipid Nanoparticles (LNP), N-acetylgalactosamine (GalNAc) conjugates, and physical methods employing novel nucleic acid formats like circular single-stranded DNA (CssDNA). Each platform possesses distinct characteristics, performance metrics, and optimal application contexts. We objectively compare their mechanisms, experimental outcomes, and practical implementation requirements to inform research and development decisions in the field of somatic cell gene therapy.
Performance Comparison of Major Non-Viral Platforms
The table below provides a quantitative comparison of the three primary non-viral delivery platforms based on current research and approved therapeutics.
Table 1: Performance Comparison of Key Non-Viral Vector Platforms
| Feature | Lipid Nanoparticles (LNP) | GalNAc Conjugates | Physical Methods (e.g., CssDNA Electroporation) |
|---|---|---|---|
| Primary Mechanism | Pseudo-lipoprotein complex; ionizable lipid-mediated endosomal escape [16] | Covalent conjugate targeting the asialoglycoprotein receptor (ASGPR) [16] | Electroporation to deliver DNA templates (e.g., CssDNA) into cells [17] |
| Typical Cargo | siRNA, mRNA, CRISPR components [15] [18] [3] | siRNA, ASO [18] [19] | Single-stranded DNA for gene insertion/correction [17] |
| Targeting Specificity | Predominantly liver (passive via ApoE/LDLR; active targeting possible) [16] | Highly specific to hepatocytes via ASGPR [19] [16] | Dependent on cell type being electroporated (ex vivo) [17] |
| Administration Route | Intravenous [16] | Subcutaneous [16] | Ex vivo delivery to cells [17] |
| Key Efficiency Metrics | ~1-4% endosomal escape efficiency [16]; >90% encapsulation efficiency [16] | <1 minute hepatocyte uptake post-SC injection [16]; High avidity (KD ≈ 5 nM) [16] | Up to 51% gene knock-in efficiency in HSPCs; 3-5x higher than linear ssDNA [17] |
| Approved Therapeutics | Patisiran (Onpattro) [1] [3], COVID-19 mRNA vaccines [18] | Givosiran (Givlaari), Lumasiran (Oxlumo), Inclisiran (Leqvio) [1] [3] | None approved to date (primarily in pre-clinical and research stage) [17] |
Platform Deep Dive: Mechanisms, Workflows, and Experimental Data
Lipid Nanoparticles (LNP)
LNPs are multi-component systems typically composed of an ionizable lipid, phospholipid, cholesterol, and a PEGylated lipid [16]. The ionizable lipid is crucial for both encapsulating the nucleic acid cargo during formulation and for mediating endosomal escape after cellular uptake. The mechanism involves the ionizable lipid's protonation in the acidic environment of the endosome, which promotes a transition to a cone-shaped morphology. This change induces membrane curvature and fusion pore formation, facilitating the release of the genetic payload into the cytosol [16].
Table 2: Key Research Reagents for LNP Formulation and Testing
| Reagent / Material | Function |
|---|---|
| Ionizable Lipids (e.g., DLin-MC3-DMA, SM-102) | Core functional lipid that encapsulates nucleic acids and mediates endosomal escape [16]. |
| Helper Phospholipid (e.g., DSPC) | Provides structural integrity to the nanoparticle bilayer [16]. |
| Cholesterol | Enhances bilayer stability and fluidity, improving nanoparticle longevity [16]. |
| PEGylated Lipid | Shields the LNP surface, reduces aggregation, and modulates pharmacokinetics [16]. |
| Microfluidic Mixer | Essential equipment for controlled, reproducible LNP formation with high encapsulation efficiency [16]. |
Figure 1: LNP Delivery Mechanism. Following intravenous administration, LNPs are coated with ApoE, enabling uptake via LDL receptor-mediated endocytosis. In the acidic endosome, ionizable lipids protonate, facilitating endosomal escape and cytosolic release of the therapeutic cargo.
GalNAc Conjugates
GalNAc conjugates represent a minimalist approach by forgoing a complex particulate carrier. Instead, a tri-antennary N-acetylgalactosamine (GalNAc) ligand is covalently conjugated directly to the therapeutic oligonucleotide (e.g., siRNA) [16]. This ligand has extremely high affinity for the asialoglycoprotein receptor (ASGPR), which is abundantly and almost exclusively expressed on the surface of hepatocytes [19] [16]. Upon binding, the conjugate is rapidly internalized via receptor-mediated endocytosis. While a significant portion of the cargo may be degraded in lysosomes, the highly efficient and repetitive cycling of ASGPR to the cell surface ensures that a sufficient fraction of the siRNA escapes into the cytoplasm to achieve potent and durable gene silencing [16].
Experimental Protocol: Rational Design of GalNAc-Lipids for CRISPR Delivery A 2023 Nature Communications study detailed the structure-guided design of GalNAc-lipids for LDLR-independent hepatic delivery of CRISPR base editors [19]. The workflow is as follows:
- Ligand Design: Two trivalent GalNAc ligand scaffolds (TRIS-based "Design 1" and lysine-based "Design 2") were synthesized and covalently attached to different lipid anchors (e.g., DSG, Chol, C20) via PEG linkers of varying lengths [19].
- Formulation: GalNAc-lipids were mixed with other lipid excipients (ionizable lipid, phospholipid, cholesterol, PEG-lipid) prior to LNP formation to ensure uniform surface display of the ligand. The LNPs were loaded with ABE8.8 mRNA and a guide RNA targeting Angptl3 or Pcsk9 [19].
- In Vivo Screening: Formulations were screened in Ldlr-/- mice to assess potency independent of the LDLR pathway. Doses ranged from 0.1 to 0.3 mg/kg [19].
- Optimization: The optimal formulation (GalNAc-Lipid "GL6" with a 36-unit PEG linker and a DSG lipid anchor at 0.05 mol%) was advanced into non-human primate (NHP) studies, including a novel LDLR-deficient NHP model [19].
- Outcome Measurement: Efficacy was measured via targeted amplicon sequencing of liver DNA to calculate editing percentages, and by ELISA to quantify target protein (ANGPTL3) reduction in blood [19].
Key Results: The optimized GalNAc-LNP (GL6) increased liver editing in LDLR-deficient NHPs from 5% (untargeted LNP) to 61%, with minimal off-target editing. In wild-type NHPs, a single dose led to a durable 89% reduction in blood ANGPTL3 protein six months post-dosing [19].
Physical Methods and Advanced Nucleic Acid Formats
Physical methods, such as electroporation, facilitate the direct delivery of nucleic acids into cells by creating transient pores in the cell membrane. The efficacy of this approach is now heavily influenced by the format of the genetic cargo. Recent advances highlight the superiority of circular single-stranded DNA (CssDNA) over traditional linear DNA templates for gene editing applications in primary cells [17].
Experimental Protocol: CssDNA for Gene Insertion in Hematopoietic Stem and Progenitor Cells (HSPCs) A 2025 Nature Communications study established a protocol for TALEN-mediated gene insertion in HSPCs using CssDNA [17]:
- Cell Culture: Human HSPCs are thawed and expanded for two days in cytokine-supplemented media [17].
- Nuclease Delivery: On day 2, cells are electroporated with mRNA encoding TALENs and helper proteins (HDR-Enh01, Via-Enh01) [17].
- Template Delivery: On day 3, cells undergo a second electroporation with the CssDNA donor template (0.6 kb to 2.2 kb in length, flanked by ~300 bp homology arms) [17].
- Analysis: Cells are analyzed on day 4 (or later) for gene knock-in efficiency (via flow cytometry for a reporter gene), cell viability, and differentiation potential (via colony-forming unit assays). Engraftment capacity is tested by transplanting edited HSPCs into immunodeficient NCG mice [17].
Key Results: The CssDNA editing process achieved over 40% gene knock-in efficiency in HSPCs, a 3- to 5-fold increase compared to linear ssDNA (LssDNA). This correlated with a higher knock-in/knock-out ratio, indicating more precise integration. Critically, CssDNA-edited HSPCs showed a higher propensity to engraft and maintain gene edits in murine models compared to AAV-edited cells, attributed to a more primitive, quiescent metabolic state [17].
Figure 2: CssDNA Workflow for HSPC Editing. The process involves sequential electroporation of nuclease mRNA and CssDNA donor template into expanded HSPCs, followed by in vitro and in vivo functional analysis.
The choice between LNP, GalNAc conjugates, and physical methods like CssDNA electroporation is dictated by the therapeutic application. LNPs offer versatility in cargo type and are the platform of choice for mRNA delivery and vaccines. GalNAc conjugates provide an elegant, highly targeted solution for liver-specific siRNA delivery, enabling subcutaneous, long-acting dosing regimens. CssDNA-based physical methods present a powerful and potentially safer alternative to AAV for ex vivo gene editing, particularly in sensitive primary cells like HSPCs, where high knock-in efficiency and preservation of cell fitness are paramount.
The ongoing refinement of these platforms—through the design of novel ionizable lipids, optimized targeting ligands, and improved nucleic acid formats—will continue to expand the reach of non-viral gene therapy, potentially moving it beyond rare diseases to address more common conditions.
In the field of gene therapy, the selection of an appropriate gene delivery vector is a fundamental determinant of therapeutic success. Vectors are engineered systems designed to transport genetic cargo into target cells and must overcome multiple biological barriers to achieve effective transduction—the process of delivering and expressing a transgene. The core properties defining any vector's utility are its transduction efficiency (the proportion of cells that successfully express the transgene), cargo capacity (the size of the genetic material it can carry), and tropism (its specificity for particular cell types) [20] [21]. These properties directly impact the safety, efficacy, and practical application of gene therapies for both research and clinical use.
Vectors are broadly categorized into viral and non-viral systems. Viral vectors leverage the natural ability of viruses to infect cells, while non-viral vectors use synthetic or physical methods for gene delivery [20] [22]. This guide provides an objective, data-driven comparison of these platforms, focusing on their key biological properties, to inform researchers and drug development professionals in their experimental and therapeutic designs.
Comparative Analysis of Vector Properties
The table below summarizes the quantitative and qualitative characteristics of major viral and non-viral vector systems, providing a clear comparison of their performance across key parameters.
Table 1: Comparative Properties of Viral and Non-Viral Vector Systems
| Vector Type | Transduction Efficiency | Cargo Capacity | Primary Tropism & Targeting Mechanism | Gene Expression Duration | Key Advantages | Key Limitations |
|---|---|---|---|---|---|---|
| Adeno-Associated Virus (AAV) | High in vivo [1] | ~4.7 kb [23] [24] | Broad; naturally occurring and engineered serotypes target muscle, liver, CNS, retina [1] [23] | Long-term, but transient in dividing cells (non-integrating) [25] | Favorable safety profile, low immunogenicity [1] [25] | Limited cargo size, potential for pre-existing immunity [26] [22] |
| Lentivirus (LV) | High ex vivo and in vivo [1] | ~8 kb [21] | Broad; can transduce dividing and non-dividing cells; pseudotyping with VSV-G expands tropism [21] [24] | Long-term (integrating) [25] [24] | Stable genomic integration, suitable for ex vivo cell therapy (e.g., CAR-T) [21] [24] | Risk of insertional mutagenesis, though reduced with SIN designs [26] [24] |
| Adenovirus (AdV) | High [1] | Up to ~36 kb [27] [24] | Very broad; naturally tropic for respiratory epithelial cells [1] | Transient (non-integrating) [20] [27] | Very large cargo capacity, high titer production [27] [22] | Strong immune response, high immunogenicity [20] [21] |
| Gamma-Retrovirus (γRV) | High for dividing cells [21] | ~10 kb [26] [24] | Restricted to dividing cells [21] [25] | Long-term (integrating) [25] | Long-term gene expression [25] | Only transduces dividing cells; higher risk of insertional mutagenesis [26] [24] |
| Lipid Nanoparticles (LNPs) | Lower than viral vectors in vivo; improving [20] [22] | High (theoretically unlimited) [22] | Primarily hepatic after systemic administration; targetability is an area of active research [1] [22] | Transient [22] | Large cargo capacity, good safety profile, scalable manufacturing [27] [22] | Lower transfection efficiency, potential toxicity at high doses, often requires repeated administration [20] [22] |
| Cationic Polymers (e.g., PEI) | Variable; generally lower than viral vectors [25] | High [25] | Low innate specificity; can be modified with targeting ligands [25] | Transient [27] | Biodegradable versions available, high cation charge density for nucleic acid compaction [27] [25] | Can be cytotoxic (e.g., non-biodegradable PEI), low specificity and transfection efficiency in vivo [25] |
Experimental Data and Performance Metrics
Quantifying Transduction Efficiency
Transduction efficiency is typically measured as the percentage of cells that successfully express the transgene post-delivery. In clinical manufacturing, this is a Critical Quality Attribute (CQA). For example, in CAR-T cell production, lentiviral transduction efficiencies typically range between 30–70%, as assessed by flow cytometry for surface marker detection or quantitative PCR for Vector Copy Number (VCN) analysis [21].
Key process parameters that critically influence transduction efficiency include:
- Cell Quality and Activation State: Pre-activating T cells upregulates viral receptor expression, significantly enhancing transduction [21].
- Multiplicity of Infection (MOI): This ratio of viral particles to target cells must be carefully titrated. A higher MOI can increase efficiency but also raises the risk of cellular toxicity and higher VCN [21].
- Transduction Enhancers: Additives such as polycations (e.g., protamine sulfate) and the use of physical methods like spinoculation (centrifugation of vector onto cells) enhance cell-vector contact and improve efficiency [21].
- Vector Engineering: Pseudotyping lentiviral vectors with the Vesicular Stomatitis Virus G-glycoprotein (VSV-G) confers broad tropism and improves stability, enhancing transduction across diverse immune cell types [21].
Assessing Cargo Capacity and Its Impact
Cargo capacity directly dictates the therapeutic genes a vector can deliver. The inherent limitations of each platform can be overcome through engineering:
- Adenoviruses offer the largest native capacity, with high-capacity "gutless" vectors accommodating up to ~36 kb of foreign DNA, enabling the delivery of large genetic sequences like the dystrophin gene for Duchenne muscular dystrophy [20] [27].
- AAV's limited ~4.7 kb capacity is a significant constraint. To overcome this, researchers use dual-AAV approaches, splitting a large transgene into two halves that recombine inside the target cell. This strategy has been successfully applied in a first-in-human therapy for hereditary hearing loss [1].
- Non-viral vectors like LNPs and polymers have a high and flexible cargo capacity, easily accommodating large CRISPR-Cas9 systems or multiple genetic elements [22].
Engineering and Measuring Tropism
Tropism defines a vector's specificity for a particular cell or tissue type. It is a critical factor for maximizing on-target delivery and minimizing off-target effects and toxicity [23]. Both viral and non-viral platforms are actively engineered to achieve precise targeting.
Diagram 1: Strategies for Engineering Viral Vector Tropism
For non-viral vectors, tropism is achieved through functionalization. This involves conjugating targeting ligands—such as antibodies, peptides, or aptamers—to the surface of nanoparticles or polymeric carriers. These ligands bind to receptors overexpressed on specific target cells, enabling receptor-mediated uptake [22]. For instance, folate-conjugated liposomes are designed to target cancer cells with high folate receptor expression [22].
Experimental validation of successful targeting involves:
- In vitro binding assays using cell lines expressing the target receptor.
- Biodistribution studies in animal models, quantifying vector genomes in different tissues after administration.
- Histological analysis of tissues to confirm transgene expression in the target cell type.
Key Experimental Protocols
Protocol: Optimizing Lentiviral Transduction of T Cells for CAR-T Therapy
This protocol outlines the critical steps for achieving high-efficiency transduction of human T cells, a cornerstone of CAR-T cell therapy manufacturing [21].
1. T Cell Activation:
- Isolate peripheral blood mononuclear cells (PBMCs) from a leukapheresis product.
- Activate T cells using anti-CD3/CD28 antibodies (e.g., Dynabeads) in a serum-free medium.
- Culture for 24-48 hours before transduction. Critical Parameter: Cell activation state directly correlates with transduction efficiency.
2. Vector Preparation and Transduction:
- Thaw a high-titer lentiviral vector (e.g., VSV-G pseudotyped) on ice.
- Calculate the required vector volume based on the Multiplicity of Infection (MOI) and cell count. An MOI between 5 and 20 is typical, but requires empirical optimization.
- Add the vector to the cells. Include transduction enhancers such as protamine sulfate (4-8 µg/mL) or Vectofusin-1 to the culture medium.
- Use Spinoculation: Centrifuge the cell-vector mixture at 800-2000 x g for 30-120 minutes at 32°C. This significantly enhances cell-vector contact and efficiency.
3. Post-Transduction Culture and Analysis:
- After 24 hours, remove the vector-containing medium and replace with fresh medium supplemented with cytokines (e.g., IL-2 at 50-100 IU/mL).
- Expand cells for 7-14 days.
- Analyze Transduction Efficiency: At day 3-5, analyze by flow cytometry for CAR or reporter gene expression.
- Measure Vector Copy Number (VCN): Use droplet digital PCR (ddPCR) on genomic DNA to ensure VCN remains below 5 copies per cell for clinical safety [21].
Protocol: Formulating and Testing Ligand-Targeted Lipid Nanoparticles (LNPs)
This protocol describes the creation of targeted LNPs for cell-specific gene delivery [22].
1. Lipid Mixture Preparation:
- Prepare a lipid mixture in ethanol. A standard formulation includes:
- Ionizable cationic lipid (e.g., DLin-MC3-DMA): For nucleic acid complexation and endosomal escape.
- Phospholipid (e.g., DSPC): Provides structural integrity.
- Cholesterol: Stabilizes the LNP bilayer.
- PEG-lipid (e.g., DMG-PEG2000): Reduces nonspecific uptake and improves stability.
- For targeting, replace a portion of the DMG-PEG2000 with a PEG-lipid conjugate bearing a targeting ligand (e.g., an antibody, peptide, or aptamer).
2. LNP Formation via Microfluidics:
- Mix the ethanolic lipid stream with an aqueous stream containing the mRNA or DNA payload at a specific flow rate ratio in a microfluidic device.
- This process results in the spontaneous formation of LNPs encapsulating the genetic material.
- Dialyze or use tangential flow filtration (TFF) to remove ethanol and exchange the buffer.
3. In Vitro Validation of Targeting:
- Incute the formulated LNPs with two cell types: one expressing the target receptor and a control cell line lacking the receptor.
- After incubation, measure transfection efficiency by quantifying reporter protein expression (e.g., GFP via flow cytometry or luciferase activity).
- To confirm receptor-specific uptake, perform a competition assay by pre-incubating the target cells with an excess of free ligand, which should block LNP binding and reduce transfection.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Vector Research and Development
| Reagent / Solution | Function & Application | Example Uses |
|---|---|---|
| VSV-G Envelope Plasmid | Pseudotyping enveloped viral vectors (LV, γRV) to confer broad tropism and enhance vector stability [21]. | Production of lentiviral vectors with high titer and ability to transduce a wide range of cell types. |
| Transduction Enhancers (e.g., Protamine Sulfate, Vectofusin-1) | Polycations that reduce electrostatic repulsion between viral particles and cell membranes, increasing transduction efficiency [21]. | Added during ex vivo transduction of T cells or hematopoietic stem cells to improve gene transfer rates. |
| Ionizable Cationic Lipids (e.g., DLin-MC3-DMA) | A component of LNPs that binds nucleic acids at low pH and promotes endosomal escape following cellular uptake [22]. | Formulating LNPs for siRNA (Onpattro) or mRNA delivery; critical for efficient cytosolic delivery. |
| Cytokines (e.g., IL-2, IL-7, IL-15) | Support the survival, expansion, and function of immune cells during and after ex vivo transduction [21]. | Culture medium supplement for manufacturing CAR-T cells and NK cells to maintain viability and potency. |
| Anti-CD3/CD28 Antibodies | Synthetic activation signals for T cells, mimicking antigen presentation and initiating cell proliferation [21]. | In vitro activation of primary T cells prior to viral transduction, a critical step for high efficiency. |
| Aurein 3.2 | Aurein 3.2, MF:C82H138N22O21, MW:1768.1 g/mol | Chemical Reagent |
| FGFR1 inhibitor-9 | FGFR1 inhibitor-9, MF:C27H20ClNO5, MW:473.9 g/mol | Chemical Reagent |
The choice between viral and non-viral vectors is a fundamental strategic decision in gene therapy research and development. As the data demonstrates, viral vectors (AAV, LV, AdV) currently offer superior transduction efficiency and have enabled multiple approved therapies, but they are constrained by immunogenicity, cargo limits, and complex manufacturing [20] [1] [25]. In contrast, non-viral vectors (LNPs, polymers) provide advantages in safety, cargo flexibility, and scalability, though they are still catching up in delivery efficiency and targeting specificity [27] [22].
The future of the field lies in the continued engineering of both platforms to overcome their limitations. For viral vectors, this involves sophisticated capsid and envelope engineering to de-target innate immunity and re-target desired tissues [23]. For non-viral vectors, the focus is on developing novel materials and conjugation strategies to enhance stability, promote endosomal escape, and achieve precise cell-specific targeting [22]. The ongoing convergence of these platforms—such as using viral components to enhance non-viral systems—promises to yield a new generation of versatile, safe, and highly effective vectors for gene therapy.
Gene therapy represents a transformative approach in modern medicine, enabling the treatment of diseases at their genetic root cause. The clinical success of these therapies is fundamentally dependent on the vectors used to deliver therapeutic genetic material into target cells. These vectors are broadly classified into viral and non-viral systems, each with distinct biological properties, clinical applications, and regulatory profiles [1]. Viral vectors, engineered from modified viruses, leverage natural infection mechanisms for high-efficiency delivery. In contrast, non-viral vectors use synthetic or biological compounds, such as lipids or polymers, to ferry genetic cargo with a typically improved safety profile [25]. As of mid-2025, the global regulatory landscape has seen the approval of at least 35 vector-based gene therapies, with 29 being viral-based and 6 utilizing non-viral methods [1] [3]. This guide provides an objective comparison of these approved therapies, detailing their performance, supported by experimental data and methodologies, to inform researchers, scientists, and drug development professionals.
The gene therapy market has witnessed accelerated growth, with 2024 being a landmark year featuring several "first" approvals, including the first T-cell receptor (TCR) therapy and the first mesenchymal stem cell product in the U.S. [28]. The table below summarizes the quantitative approval landscape for vector-based gene therapies.
Table 1: Global Approval Summary for Vector-Based Gene Therapies (as of 2025)
| Vector Category | Number of Approved Therapies | Key Examples (Indication) |
|---|---|---|
| Viral-Based Total | 29 [1] | Kymriah (Leukemia) [1], Zolgensma (SMA) [29], Luxturna (Retinal Dystrophy) [1] |
| Lentivirus (LV) | 14 (ex vivo) [1] | Zynteglo (Beta Thalassemia) [1], Skysona (Cerebral Adrenoleukodystrophy) [1] |
| Adenovirus (Ad) | 4 [1] | Gendicine (Head and Neck Cancer) [1], Adstiladrin (Bladder Cancer) [1] |
| Adeno-associated Virus (AAV) | 9 [1] | Hemgenix (Hemophilia B) [1], Elevidys (DMD) [1], Roctavian (Hemophilia A) [1] |
| Non-Viral-Based Total | 6 [1] | Onpattro (hATTR Amyloidosis) [1], Givlaari (Acute Hepatic Porphyria) [1] |
| Lipid Nanoparticle (LNP) | 1 [1] | Onpattro (siRNA for hATTR) [1] [3] |
| GalNAc Conjugation | 5 [1] | Leqvio (Hypercholesterolemia) [1], Oxlumo (Primary Hyperoxaluria) [1] |
The distribution of approved products underscores the historical dominance of viral vectors, particularly in ex vivo cell engineering and in vivo treatments for monogenic diseases. However, the recent rise of non-viral platforms, especially GalNAc-conjugated therapies for liver-targeted delivery, highlights a significant shift towards scalable and less immunogenic delivery solutions [3].
Approved Viral Vector Therapies: In-Depth Analysis
Viral vectors are the cornerstone of approved gene therapies, prized for their high transduction efficiency and ability to achieve long-term gene expression [25]. The choice of viral vector is dictated by the therapeutic goal, target cell type, and required duration of expression.
Table 2: Key Approved Viral Vector Therapies and Clinical Evidence
| Product (Vector) | Therapeutic Indication | Key Clinical Trial Efficacy Data | Administration Route & Rationale |
|---|---|---|---|
| Kymriah (LV) [1] | Acute Lymphoblastic Leukemia | Pivotal trial (ELIANA): 83% overall remission rate in r/r pediatric patients [1]. | Ex vivo transduction of patient T-cells ensures targeted delivery to cancerous B-cells, minimizing systemic exposure. |
| Zolgensma (AAV9) [29] | Spinal Muscular Atrophy (SMA) | Phase 3 STR1VE trial: 100% event-free survival at 14 months; 92% achieving motor milestones unseen in natural history [29]. | Intravenous (IV) systemic delivery. AAV9 serotype has high tropism for motor neurons, crossing the blood-brain barrier. |
| Luxturna (AAV2) [1] | RPE65-mediated Retinal Dystrophy | Phase 3: Significant improvement in multi-luminance mobility test; 93% of participants showed improved functional vision [1]. | Subretinal injection. Localized delivery ensures high vector concentration at the target retinal pigment epithelium cells. |
| Itvisma (AAV9) [30] | Spinal Muscular Atrophy (SMA) in patients ≥2 years | Phase 3 STEER study: Statistically significant improvement in motor function (HFMSE score) sustained over 52 weeks [30]. | Intrathecal injection. Direct delivery to cerebrospinal fluid allows lower vector dose and targets spinal cord motor neurons. |
Experimental Protocols for Viral Therapies
The development and validation of these therapies rely on standardized preclinical and clinical protocols.
Ex Vivo Lentiviral Protocol (e.g., CAR-T Production): Patient T-cells are harvested via leukapheresis and activated ex vivo using anti-CD3/anti-CD28 beads. These activated T-cells are then transduced with a recombinant lentiviral vector encoding the chimeric antigen receptor (CAR) in the presence of polycations like polybrene to enhance transduction efficiency. The transduced cells are expanded in culture using recombinant human cytokines (e.g., IL-2) for 7-10 days before being infused back into the patient [1] [24].
In Vivo AAV Protocol (e.g., ITVSMA Clinical Trial): The Phase III STEER study for Itvisma was a multicenter, randomized, controlled trial. Patients with confirmed SMN1 mutations received a single intrathecal bolus injection of the AAV9-based therapy. The primary efficacy endpoint was the change from baseline in the Hammersmith Functional Motor Scale-Expanded (HFMSE) score at 52 weeks. Safety was assessed by monitoring for adverse events of special interest, including hepatotoxicity and cardiotoxicity, consistent with the known risks of AAV therapies [30].
Approved Non-Viral Vector Therapies: In-Depth Analysis
Non-viral vectors offer advantages in safety, manufacturing scalability, and reduced immunogenicity. Their approved applications are pioneering targeted delivery, particularly to the liver.
Table 3: Key Approved Non-Viral Vector Therapies and Clinical Evidence
| Product (Platform) | Therapeutic Indication | Key Clinical Trial Efficacy Data | Mechanism of Action |
|---|---|---|---|
| Onpattro (LNP) [1] [3] | Hereditary Transthyretin Amyloidosis (hATTR) | APOLLO Phase 3: Significantly reduced modified Neuropathy Impairment Score +7 points vs. placebo. | Delivers siRNA to hepatocytes, mediating degradation of mutant and wild-type TTR mRNA. |
| Givlaari (GalNAc) [1] | Acute Hepatic Porphyria (AHP) | Phase 3 ENVISION: Reduced porphyria attacks by 74% compared to placebo. | GalNAc ligand targets asialoglycoprotein receptor (ASGPR) on hepatocytes. Delivers siRNA against aminolevulinic acid synthase 1 (ALAS1). |
| Leqvio (GalNAc) [1] | Hypercholesterolemia | Phase 3 ORION-9,10,11: Sustained reduction of LDL cholesterol by ~50% with biannual dosing. | GalNAc-conjugated siRNA targets and silences PCSK9 mRNA in hepatocytes. |
| Rivfloza (GalNAc) [1] | Primary Hyperoxaluria | Phase 3: Achieved substantial reduction in urinary oxalate levels. | GalNAc-conjugated siRNA against hydroxyacid oxidase 1 (HAO1) gene, reducing oxalate production. |
Experimental Protocols for Non-Viral Therapies
The clinical validation of non-viral therapies involves unique considerations for biodistribution and silencing efficacy.
LNP-siRNA Protocol (e.g., Onpattro Clinical Trial): The APOLLO trial was a randomized, double-blind, placebo-controlled Phase 3 study. Patients received 0.3 mg/kg of patisiran via intravenous (IV) infusion every three weeks. Co-administration of corticosteroids, acetaminophen, and H1/H2 blockers was required to preempt infusion-related reactions. The primary endpoint was the change from baseline in the mNIS+7 score at 18 months. Efficacy was corroborated by measuring serum TTR protein levels as a pharmacodynamic biomarker [1] [3].
GalNAc-siRNA Protocol (e.g., Givlaari Clinical Trial): The ENVISION trial was a randomized, double-blind, placebo-controlled Phase 3 study. Patients received subcutaneous injections of givosiran at a dose of 2.5 mg/kg monthly. The primary efficacy endpoint was the annualized rate of composite porphyria attacks. The delivery platform's success was evidenced by measuring urinary aminolevulinic acid levels, confirming target engagement in the heme biosynthesis pathway [1].
Comparative Analysis: Performance Data of Viral vs. Non-Viral Vectors
The choice between viral and non-viral vectors involves a careful trade-off between efficacy, safety, and manufacturability.
Diagram Title: Vector selection is a multi-parameter decision tree.
Table 4: Direct Comparison of Viral vs. Non-Viral Vector Properties
| Parameter | Viral Vectors (AAV, LV) | Non-Viral Vectors (LNP, GalNAc) |
|---|---|---|
| Transduction/Efficiency | High. Natural infection mechanism; sustained expression [25]. | Variable, often lower. Must overcome endosomal degradation; typically transient effect [25]. |
| Cargo Capacity | Limited. AAV: ~4.7kb [3]. LV: ~8kb [24]. | Large. Can deliver large DNA, mRNA, CRISPR ribonucleoproteins [25]. |
| Immunogenicity | Moderate to High. Can trigger innate/adaptive immunity, limiting re-dosing [25] [3]. | Low. Safer profile, but LNPs can cause infusion reactions [25] [3]. |
| Genotoxicity Risk | Yes. LV/RV integrate into host genome, risk of insertional mutagenesis [24] [3]. | Very Low. Primarily episomal; no known risk of insertional mutagenesis [25]. |
| Manufacturing & Scalability | Complex and costly. Requires production in mammalian cell lines [25]. | Simpler and more scalable. Based on synthetic chemistry, good for GMP [25]. |
| Typical Dosing | Often one-time (e.g., Zolgensma, Luxturna) [1] [29]. | Often multiple doses (e.g., Leqvio is biannual) [1]. |
The Scientist's Toolkit: Key Research Reagents & Materials
Advancing gene therapy research requires a suite of specialized reagents and materials. The table below details essential tools for developing and evaluating vector systems.
Table 5: Essential Research Reagents for Vector Development
| Research Reagent / Material | Primary Function | Application Example |
|---|---|---|
| Polycations (e.g., Polybrene) | Enhance viral vector infection efficiency by neutralizing charge repulsion between vector and cell membrane [24]. | Used during ex vivo transduction of T-cells with lentiviral vectors to improve CAR transfer efficiency [24]. |
| LNP Formulation Lipids | Cationic/ionizable lipids bind nucleic acids; PEG-lipids stabilize particles; helper lipids support bilayer structure [25]. | Formulate siRNA (as in Onpattro) or mRNA into nanoparticles for in vivo delivery [1] [25]. |
| Cytokines (e.g., IL-2) | T-cell growth factor that promotes the expansion and survival of genetically modified T-cells during ex vivo culture [24]. | Critical for expanding CAR-T cells to sufficient numbers for therapeutic infusion after transduction [24]. |
| AAV Serotype Libraries | Different AAV serotypes (e.g., AAV2, AAV8, AAV9) exhibit distinct tropisms for various tissues (e.g., liver, CNS, muscle) [25] [3]. | Screening to identify the optimal serotype for a specific target tissue in preclinical models [3]. |
| GalNAc Ligand | A carbohydrate ligand that binds with high affinity to the ASGPR, which is highly expressed on hepatocytes [1] [3]. | Conjugated to siRNA or ASO therapeutics to achieve targeted delivery to the liver [1]. |
| Selective Promoters/Enhancers | Regulatory DNA sequences that restrict transgene expression to specific cell or tissue types (e.g., synapsin promoter for neurons) [3]. | Incorporated into viral vectors (AAV, LV) to limit off-target expression and enhance safety [3]. |
| PAN endonuclease-IN-2 | PAN endonuclease-IN-2|Influenza Antiviral Research Compound | PAN endonuclease-IN-2 is a potent research compound that inhibits influenza virus replication. For Research Use Only. Not for human or veterinary diagnostic or therapeutic use. |
| Cyclo(Gly-Arg-Gly-Glu-Ser-Pro) | Cyclo(Gly-Arg-Gly-Glu-Ser-Pro), MF:C23H37N9O9, MW:583.6 g/mol | Chemical Reagent |
Diagram Title: LNP and viral vectors have distinct production workflows.
The current market landscape demonstrates a dynamic and maturing gene therapy sector. Viral vectors, particularly AAV and LV, have established a stronghold for indications requiring high-efficiency, durable gene expression, as evidenced by landmark therapies for SMA, retinal diseases, and blood cancers. Meanwhile, non-viral vectors, led by LNPs and GalNAc platforms, are carving out a critical niche by offering a safer, more scalable, and commercially viable path forward, especially for liver-targeted diseases and applications requiring redosability. The future of gene delivery lies not in a single dominant platform, but in the continued innovation and refinement of both viral and non-viral systems. Emerging strategies like capsid engineering, dual-vector systems to overcome cargo limits, and advanced non-viral designs for extrahepatic delivery will collectively expand the reach of gene therapies to a broader spectrum of human diseases [1] [3].
Therapeutic Applications and Clinical Translation of Vector Platforms
Lentiviral vectors (LVs) have emerged as a premier delivery platform for ex vivo gene therapy, enabling the genetic modification of a patient's own cells outside the body before reinfusion. Derived from the human immunodeficiency virus (HIV-1), LVs are distinguished by their ability to transduce both dividing and non-dividing cells and provide stable genomic integration and long-term transgene expression [24] [31]. These properties make LVs particularly suitable for engineering hematopoietic stem cells (HSCs) and T lymphocytes, the cornerstone of advanced therapies for genetic disorders and cancers [24] [31] [32]. This guide objectively compares LV performance in two dominant applications: CAR-T cell therapies for hematological malignancies and HSC-based therapies for inherited monogenic diseases, providing researchers with a direct comparison of experimental data, protocols, and safety considerations.
Comparative Analysis of LV Performance in CAR-T vs. HSC Therapies
The performance of LV systems differs significantly between therapeutic applications due to distinct biological targets and clinical requirements. The table below summarizes key performance metrics and their direct impact on therapeutic outcomes.
| Performance Metric | LV in CAR-T Cell Therapy | LV in HSC-Based Therapy | Impact on Therapeutic Outcome |
|---|---|---|---|
| Typical Transduction Efficiency | 30-70% [31] | Varies; can be optimized via pre-stimulation & enhancers [31] | Directly influences the dose of therapeutic cells; critical for product potency. |
| Vector Copy Number (VCN) Safety Window | Generally maintained below 5 copies/cell [31] | Carefully monitored; clonal expansions reported (e.g., in HMGA2) [33] | High VCN raises genotoxicity risk; must balance with sufficient transgene expression. |
| Key Challenge | Functional persistence of CAR-T cells; tonic signaling [34] | Ensuring engraftment and long-term polyclonal reconstitution [35] [33] | CAR-T: Impacts durability of anti-tumor response.HSC: Affects cure longevity and safety profile. |
| Primary Safety Concern | Secondary malignancies (rare reports) [33] | Insertional mutagenesis leading to clonal dominance or malignancy [33] | Drives requirement for extensive integration site analysis and long-term patient follow-up. |
Detailed Experimental Protocols and Workflows
Ex Vivo Manufacturing of LV-Modified CAR-T Cells
The production of CAR-T cells is a multi-step process where critical process parameters (CPPs) must be tightly controlled to ensure the critical quality attributes (CQAs) of the final product [31].
- Leukapheresis and T-Cell Isolation: T cells are collected from the patient via leukapheresis and isolated from other blood components [31].
- T-Cell Activation: Isolated T cells are activated using methods such as CD3/CD28 stimulation, a critical step that upregulates receptors for viral entry and primes cells for transduction [31].
- Lentiviral Transduction:
- The activated T cells are exposed to the LV carrying the CAR transgene.
- Multiplicity of infection (MOI), which defines the ratio of functional vector particles to target cells, is carefully titrated to balance high transduction efficiency against cell toxicity and excessive VCN [31]. MOI is a key CPP.
- Spinoculation (centrifugation during transduction) is often employed to enhance cell-vector contact and improve efficiency [31].
- Transduction enhancers (e.g., polycations like protamine sulfate) may be added to the culture medium to increase transduction yield [31].
- Ex Vivo Expansion: Transduced T cells are expanded in culture over approximately 10-14 days. The culture medium is supplemented with a cytokine cocktail (typically including IL-2) to support T-cell growth and function [31].
- Harvest and Formulation: Once sufficient cell numbers are achieved, the CAR-T cells are harvested, washed, and formulated into a final product for infusion back into the patient [31].
Ex Vivo Manufacturing of LV-Modified HSCs for Genetic Disorders
The protocol for HSC-based therapy shares similarities with CAR-T manufacturing but involves unique steps critical for engraftment.
- HSC Collection: Hematopoietic stem and progenitor cells are harvested from the patient's bone marrow or mobilized peripheral blood [35].
- Pre-Stimulation Culture: Unlike T cells, HSCs are largely quiescent. They require a period of pre-stimulation with cytokine cocktails (e.g., SCF, TPO, FLT3L) to induce cell cycle entry, which is necessary for efficient LV transduction [24].
- Lentiviral Transduction: The pre-stimulated HSCs are transduced with the LV carrying the therapeutic gene. Similar to CAR-T production, MOI optimization and the use of transduction enhancers are critical CPPs for achieving high gene marking while maintaining cell viability [31].
- Patient Conditioning: Prior to reinfusion of the gene-corrected HSCs, the patient must undergo myeloablative conditioning. This typically involves chemotherapeutic agents like busulfan (see Table 2) to create "space" in the bone marrow niche for the transplanted cells to engraft [35].
- Reinfusion and Engraftment: The transduced HSC product is infused back into the patient. Successful, polyclonal long-term engraftment is monitored over subsequent months, as this indicates stable reconstitution of a healthy hematopoietic system [35] [33].
The following workflow diagrams illustrate and contrast these two critical processes.
Safety and Genotoxicity Landscape
The genotoxicity associated with vector integration remains a primary safety consideration for all integrating vector platforms. The field has evolved from first-generation gamma-retroviral vectors (γRVs), which carried a high risk of insertional mutagenesis and leukemogenesis, to safer self-inactivating (SIN) lentiviral vectors [33] [36].
- γRV Risks: Early clinical trials for X-SCID, CGD, and WAS using γRVs resulted in a significant number of genotoxicity events, including T-cell acute lymphoblastic leukemia and myelodysplastic syndrome. These were directly linked to the transactivation of proto-oncogenes like LMO2 and MDS-EVI1 by powerful viral enhancer/promoter elements in the vector long terminal repeats (LTRs) [33].
- SIN-LV Safety Profile: SIN-LVs, which have deleted the viral promoter/enhancer from the LTR, demonstrate a safer integration profile with a reduced preference for transcription start sites, thereby lowering the risk of oncogene transactivation [24] [33]. However, genotoxic events, including clonal expansions and rare cases of malignancy, have still been reported with SIN-LVs in HSC therapies for conditions like X-linked adrenoleukodystrophy and sickle cell disease [33]. Contributing factors can include the specific design of the internal promoter, the use of certain insulator elements, and patient-specific factors like the underlying disease and conditioning regimen [33].
Rigorous vector integration site analysis and long-term patient monitoring are now mandatory components of clinical trials and approved therapies to assess and manage this risk [33].
The Scientist's Toolkit: Essential Research Reagents
Successful development and manufacturing of LV-based therapies rely on a suite of critical reagents and materials. The table below details key solutions and their functions.
| Research Reagent / Material | Critical Function in Protocol |
|---|---|
| CD3/CD28 Antibodies | Artificial antigen-presenting cell systems for robust, polyclonal T-cell activation, a prerequisite for efficient LV transduction [31]. |
| Lentiviral Vector (SIN configuration) | Delivery of the therapeutic transgene (CAR or corrective gene) with a improved safety profile due to deleted viral promoter/enhancer sequences [33]. |
| VSV-G Pseudotyped Envelope | The most common envelope protein for LVs, confers broad tropism and high stability, enabling efficient transduction of diverse immune and stem cells [31]. |
| Cytokine Cocktails (IL-2, IL-7, IL-15 for T cells; SCF, TPO, FLT3L for HSCs) | Support survival, proliferation, and maintenance of desired cell phenotypes during and after the ex vivo culture and transduction process [31]. |
| Transduction Enhancers (e.g., Protamine Sulfate) | Polycations that reduce electrostatic repulsion between viral particles and cell membranes, thereby increasing transduction efficiency [31]. |
| Myeloablative Agents (e.g., Busulfan) | Alkylating chemotherapy used in HSC therapy to ablate host bone marrow and create niche "space" for the engraftment of gene-corrected HSCs [35]. |
| Sdh-IN-10 | Sdh-IN-10||RUO |
| Shp2-IN-24 | Shp2-IN-24, MF:C23H22ClN5O4S, MW:500.0 g/mol |
Lentiviral vectors have undeniably cemented their role as a powerful and versatile tool in ex vivo gene therapy, as evidenced by their central function in approved CAR-T and HSC-based products. The choice of LV platform and the optimization of manufacturing protocols are highly application-dependent, requiring careful balancing of transduction efficiency, transgene persistence, and long-term safety. While the advent of SIN configurations has markedly improved the risk-benefit profile, the field continues to be challenged by the inherent, albeit reduced, risk of insertional mutagenesis and the complexities of scalable manufacturing. Future advancements will likely focus on further refining vector design for enhanced safety and controlled expression, optimizing point-of-care manufacturing to improve accessibility, and integrating novel genome-editing tools like CRISPR-Cas for next-generation therapeutic strategies [37] [36].
The choice of a delivery vector is a fundamental decision in the development of in vivo gene therapies. For treatments targeting monogenic and neurological disorders, the landscape is predominantly shaped by the competition between viral and non-viral strategies. Among viral vectors, Adeno-Associated Virus (AAV) has emerged as a leading platform, prized for its long-term gene expression and favorable safety profile relative to other viruses. Non-viral methods, such as lipid nanoparticles (LNPs), offer distinct advantages in scalability and reduced immunogenicity. This guide provides an objective, data-driven comparison of AAV's performance against these alternatives, focusing on its central role in advancing treatments for complex diseases of the brain and monogenic organs.
Vector Comparison: AAV, Lentivirus, and LNP at a Glance
The selection of a delivery vector involves balancing cargo capacity, persistence of expression, immunogenicity, and manufacturability. The table below summarizes the core characteristics of the primary viral and non-viral platforms for in vivo gene therapy.
Table 1: Technical Comparison of Major In Vivo Gene Delivery Platforms
| Feature | AAV (Viral) | Lentivirus (Viral) | LNP (Non-Viral) |
|---|---|---|---|
| Primary Use Case | In vivo gene replacement for CNS, eye, liver [38] | Ex vivo cell therapy (e.g., CAR-T, HSCs) [38] | Gene editing (e.g., CRISPR/mRNA), vaccines [38] |
| Cargo Capacity | ~4.7 kb (Strict) [38] [39] | ~10 kb (Moderate) [38] | Flexible / High [38] |
| Genetic Persistence | Episomal (Long-term in non-dividing cells) [38] | Integrated (Permanent in dividing cells) [38] | Transient (Ideal for editing) [38] |
| Immunogenicity | High (Pre-existing antibodies prevent re-dosing) [38] | Low (Use is mostly ex vivo) [38] | Low (Re-dosable) [38] |
| Manufacturing COGS | High (Complex cell culture & purification) [38] | High (Shear sensitivity, low yield) [38] | Low to Medium (Chemical synthesis) [38] |
| Key Bottleneck | Empty/Full capsid separation [38] | Viral stability & titer [38] | Lipid purity & microfluidic fouling [38] |
AAV in Action: Quantitative Outcomes in Disease Models
AAV's therapeutic potential is best illustrated by its efficacy in diverse preclinical and clinical studies. The following table compiles key experimental data and quantitative outcomes from recent research across neurological and monogenic disorders.
Table 2: Summary of Experimental AAV Gene Therapy Data in Preclinical and Clinical Studies
| Disease / Model | Therapeutic Transgene | AAV Serotype & Delivery | Key Efficacy Outcomes |
|---|---|---|---|
| MYBPC3 Cardiomyopathy (Murine Model) [39] | Human MYBPC3 (TN-201) | AAV9; Systemic (Retro-orbital) | Reversal of cardiac hypertrophy & systolic dysfunction; improved diastolic function; prolonged survival at dose of 3E13 vg/kg [39]. |
| Parkinson's Disease (Human Phase 1/2) [40] | Glutamic Acid Decarboxylase (GAD) | AAV2; Direct intracranial (Subthalamic Nucleus) | High dose (210B vg) group showed significant 18-point improvement in UPDRS Part III motor score off medication [40]. |
| FOXG1 Syndrome (Murine Model) [41] [42] | Human FOXG1 | AAV9; Intracerebroventricular (Postnatal) | Rescue of corpus callosum agenesis; restoration of dentate gyrus morphology; normalization of myelination [41] [42]. |
| Rett Syndrome (Human Clinical Trial) [43] | MECP2 (RETT-001) | AAV; Intrathecal | Marked improvement in motor skills, eating, and communication in a 6-year-old patient [43]. |
| Parkinson's Disease (Human Phase Ib) [44] | GDNF (AB-1005) | AAV2; Direct intracranial (Putamen) | Favorable safety profile; positive trends in clinical measures (MDS-UPDRS) and reduction in medication (LEDD) at 36 months [44]. |
Experimental Protocols for Key Applications
AAV9-Mediated Gene Replacement in Cardiomyopathy
This protocol details the methodology from a study demonstrating the reversal of cardiac dysfunction in a murine model of MYBPC3 cardiomyopathy [39].
- 1. Vector Engineering: An AAV genomic cassette was engineered for optimal packaging and cardiomyocyte expression. The TN-201 construct uses a minimized cardiac-specific promoter (pCard1) to drive the expression of the full-length human MYBPC3 cDNA (3.825 kb), fitting within the AAV size constraint [39].
- 2. Animal Model Generation: A Mybpc3 knockout murine model was established on the C57BL/6 background using CRISPR-Cas9 paired gRNA to delete exons one and two, modeling severe human hypertrophic cardiomyopathy [39].
- 3. In Vivo Delivery: Symptomatic adult mice received a single systemic injection of AAV9-TN-201 via the retro-orbital sinus at a clinically relevant dose of 3E13 vector genomes per kilogram (vg/kg) [39].
- 4. Efficacy Assessment: Animals were monitored for 12-24 weeks. Functional outcomes were measured via echocardiography to assess systolic and diastolic function. Molecular efficacy was confirmed by quantifying MYBPC3 RNA and protein levels in heart tissue, and survival was tracked as a key long-term endpoint [39].
AAV2-Mediated Neurotransmitter Modulation in Parkinson's Disease
This protocol outlines the approach for a clinical trial using AAV to modulate brain circuitry in Parkinson's disease [40].
- 1. Vector Design: An AAV2 vector was constructed to carry the glutamic acid decarboxylase (GAD) gene, which encodes the enzyme responsible for synthesizing the inhibitory neurotransmitter GABA [40].
- 2. Surgical Delivery: The gene therapy (AAV-GAD) was delivered directly and bilaterally into the patients' subthalamic nuclei (STN). This precise intracerebral injection was performed using stereotactic neurosurgery with MRI guidance to ensure accurate targeting [40].
- 3. Patient Cohort: The Phase 1/2 trial enrolled 14 participants with idiopathic Parkinson's disease and moderate motor symptoms. Patients were randomized to receive a low dose (70B vg), a high dose (210B vg), or a sham surgical procedure [40].
- 4. Outcome Measures: The primary endpoint was safety over six months. Efficacy was evaluated as the change in the Unified Parkinson's Disease Rating Scale (UPDRS) Part III motor score in a medication-off state. Quality of life was measured using the Parkinson's Disease Questionnaire (PDQ-39) [40].
Visualizing Therapeutic Mechanisms and Workflows
AAV-Mediated Gene Replacement Mechanism
The following diagram illustrates the core mechanism of AAV-mediated gene replacement therapy for a monogenic disorder like MYBPC3 cardiomyopathy [39] or FOXG1 syndrome [41].
Experimental Workflow for Preclinical AAV Evaluation
This workflow outlines the key steps for evaluating a novel AAV-based gene therapy, from vector design to in vivo assessment, as demonstrated in the cited research [39] [41].
The Scientist's Toolkit: Essential Research Reagents
Successful development and evaluation of AAV gene therapies rely on a suite of specialized reagents and tools. The following table details key solutions used in the featured experiments.
Table 3: Essential Research Reagent Solutions for AAV Gene Therapy Development
| Research Reagent / Tool | Function in Development | Example Use Case |
|---|---|---|
| AAV Serotypes (e.g., AAV2, AAV9) | Determines tissue tropism and transduction efficiency. AAV9 crosses the blood-brain barrier, enabling less invasive delivery [45] [41]. | AAV9 used for systemic delivery to target the heart [39] and brain [41]. |
| Tissue-Specific Promoters | Drives transgene expression in target cells while minimizing off-target expression, improving safety and efficacy [39]. | A minimized cardiac-specific promoter (pCard1) was engineered for high MYBPC3 expression in cardiomyocytes [39]. |
| Optimized Expression Cassette | A genetically engineered component designed to maximize therapeutic gene packaging and expression within AAV's size constraints. | The TN-201 cassette enhanced packaging and protein expression for the large MYBPC3 gene [39]. |
| Stereotactic Neurosurgery Systems | Enables precise, image-guided delivery of AAV vectors to deep brain structures for neurological disorders. | Used for convective delivery of AAV2-GDNF to the putamen in Parkinson's patients [44]. |
| Reporter Constructs (e.g., GFP) | A research tool using a gene that encodes a fluorescent protein, allowing for visualization of which cells have been successfully transduced by the therapy. | AAV9 with a GFP reporter was used to quantify transduction efficiency in iPSC-derived cardiomyocytes [39]. |
| DEALA-Hyp-YIPD | DEALA-Hyp-YIPD, MF:C50H74N10O19, MW:1119.2 g/mol | Chemical Reagent |
| Icmt-IN-12 | Icmt-IN-12|ICMT Inhibitor|For Research Use | Icmt-IN-12 is a potent ICMT inhibitor for cancer research. This product is For Research Use Only. Not for human or veterinary diagnostic or therapeutic use. |
AAV-based in vivo gene therapy has solidified its role as a powerful modality for treating monogenic and neurological disorders. Its ability to provide long-lasting gene expression in non-dividing tissues makes it uniquely suited for diseases like Parkinson's, Rett syndrome, and certain cardiomyopathies. However, the choice of vector is not one-size-fits-all. While AAV excels in these areas, its limitations—notably cargo capacity and immunogenicity—are significant. LNPs and other non-viral vectors present a compelling alternative for applications requiring transient expression, such as gene editing, or for diseases where re-dosing is a critical part of the treatment strategy. The future of the field lies in a delivery-agnostic approach, where the vector is matched to the specific therapeutic goal, and in continued innovation to overcome the current limitations of all platform technologies.
The field of gene therapy is undergoing a transformative shift, fueled by the clinical success of non-viral delivery platforms, particularly lipid nanoparticles (LNPs). While viral vectors have historically dominated clinical applications, non-viral vectors are gaining prominence due to their improved safety profiles, commercial scalability, and reduced immunogenicity [1] [3]. The development of RNA-based therapeutics faces significant hurdles, primarily inefficient tissue targeting and severe side effects, which have led to the termination of many clinical trials [46]. The breakthrough came with the approval of the first LNP-based siRNA therapeutic, Patisiran (Onpattro), in 2018, which provided a validated platform for systemic RNA delivery [1] [3]. This was followed by the global deployment of LNP-formulated mRNA vaccines during the COVID-19 pandemic, which demonstrated the robust potential of this technology on an unprecedented scale [47]. This review objectively examines the clinical performance of LNP-delivered siRNA and mRNA therapies, comparing them with viral vector alternatives and highlighting the experimental data that underpins their success.
LNP Delivery Mechanisms and Optimization Strategies
Core Mechanism of Action
Lipid nanoparticles are sophisticated multicomponent systems designed to protect RNA cargo and facilitate its intracellular delivery. The core mechanism involves several critical steps. First, LNPs encapsulate RNA molecules, shielding them from degradation by serum nucleases during systemic circulation [48]. Following administration, LNPs accumulate in target tissues, with first-generation systems showing a strong natural tropism for the liver [49]. Upon reaching the target cell, LNPs are internalized via endocytosis. The key subsequent step is endosomal escape, a process where the LNP's ionizable lipids become protonated in the acidic endosomal environment, leading to disruption of the endosomal membrane and release of the RNA into the cytosol [50]. For mRNA therapies, the released mRNA is then translated into the target protein. For siRNA therapies, the siRNA is loaded into the RNA-induced silencing complex (RISC), which guides it to the complementary mRNA target for cleavage and degradation, thereby silencing gene expression [46].
Composition and Performance Optimization
The performance of LNPs is heavily influenced by their individual lipid components, each playing a distinct role [50]. The structure-function relationships of these components are a primary focus of formulation research.
- Ionizable Lipids: These are the most critical functional component of LNPs. Their positive charge at acidic pH facilitates complexation with nucleic acids and enables endosomal escape. Research shows that tuning the pKa of ionizable lipids to approximately 6.5 is optimal for endosomal escape while minimizing cytotoxicity [50] [49].
- Helper Lipids: The choice of helper lipid, such as DOPE (dioleoylphosphatidylethanolamine) or DSPC (distearoylphosphatidylcholine), significantly impacts LNP fusogenicity and efficiency. Studies indicate that DOPE promotes non-bilayer structures that enhance endosomal escape, while DSPC provides greater stability [50]. The optimal choice depends on the RNA cargo and target tissue.
- PEG-Lipids: These lipids shield the LNP surface, reduce aggregation, control particle size, and improve pharmacokinetics by preventing rapid clearance. A critical trade-off is that high PEG content can inhibit cellular uptake and endosomal escape, which is managed by using PEG-lipids with reversible linkages [50].
The following diagram illustrates the functional structure of an LNP and its mechanism of action from cellular uptake to therapeutic effect.
Diagram 1: LNP Structure and Mechanism of Action. This figure illustrates the multicomponent structure of a lipid nanoparticle and its sequential mechanism of action from cellular uptake to the final therapeutic outcome, which differs for mRNA and siRNA cargo.
Clinical Success Stories and Comparative Performance Data
Approved LNP-based Therapies
The clinical validation of LNP platforms began with siRNA therapeutics and expanded dramatically with mRNA vaccines. The table below summarizes key approved LNP-delivered RNA therapies, detailing their indications, molecular targets, and reported clinical outcomes.
Table 1: Clinically Approved LNP-delivered siRNA and mRNA Therapies
| Therapy (Brand Name) | RNA Type | Indication | Molecular Target | Key Clinical Outcome | Approval Year |
|---|---|---|---|---|---|
| Patisiran (Onpattro) [1] [3] | siRNA | Hereditary transthyretin-mediated amyloidosis (hATTR) | Transthyretin (TTR) mRNA | ~80% reduction in serum TTR levels; improved neuropathy [1]. | 2018 |
| BNT162b2 (Comirnaty) [46] | mRNA | COVID-19 | SARS-CoV-2 Spike protein | ~95% efficacy against symptomatic COVID-19 in pivotal trial [46]. | 2020/2021 |
| mRNA-1273 (Spikevax) [46] | mRNA | COVID-19 | SARS-CoV-2 Spike protein | ~94% efficacy against symptomatic COVID-19 in pivotal trial [46]. | 2020/2021 |
Quantitative Comparison of Delivery Efficiency and Safety
A critical evaluation of LNP performance against viral vectors requires examining quantitative data on delivery efficiency, durability, and safety from preclinical and clinical studies. The following table consolidates such comparative data.
Table 2: Performance Comparison of LNP vs. Viral Vectors for RNA Delivery
| Parameter | LNP (siRNA/mRNA) | Adeno-Associated Virus (AAV) | Lentivirus (LV) |
|---|---|---|---|
| Transfection Efficiency | High in hepatocytes [3] | Very high; sustained expression in non-dividing cells [1] [25] | High; stable integration in dividing cells [25] |
| Expression Durability | Transient (days to weeks) [46] | Long-term (potentially years) [1] [25] | Long-term (stable genomic integration) [1] [25] |
| Cargo Capacity | ~10 kb (practical limit for mRNA) [48] | Limited (~4.7 kb) [3] | ~8-10 kb [25] |
| Immunogenicity | Moderate (can cause infusion reactions, manageable with premedication) [3] [48] | High; pre-existing immunity and capsid/CD8+ T-cell response are major concerns [3] [25] | Moderate; mainly used ex vivo to avoid immune response [25] |
| Risk of Insertional Mutagenesis | None (non-integrating) [25] | Low (primarily episomal) [25] | Present (integrates into host genome) [3] [25] |
| Manufacturing Scalability | High (synthetic, scalable processes) [3] [25] | Challenging (cell-based, low yields, high cost) [3] [25] | Challenging (cell-based, complex purification) [25] |
Detailed Experimental Protocols and Workflows
To provide a reproducible resource for researchers, this section outlines standard experimental methodologies for evaluating LNP performance, from formulation to in vitro and in vivo assessment.
LNP Formulation and Characterization Protocol
The standard method for LNP preparation is microfluidic mixing, which ensures reproducible, size-controlled particles [51].
Protocol: LNP Formulation via Microfluidic Mixing
- Lipid Stock Preparation: Dissolve ionizable lipid, helper lipid (DSPC or DOPE), cholesterol, and PEG-lipid in ethanol at a precise molar ratio. A typical molar ratio is 50:10:38.5:1.5 (ionizable:helper:cholesterol:PEG-lipid) [50].
- RNA Preparation: Dilute the RNA (siRNA or mRNA) in an aqueous citrate buffer (e.g., 10 mM citrate, pH 4.0).
- Mixing: Use a microfluidic device to rapidly mix the ethanolic lipid stream with the aqueous RNA stream at a defined flow rate and ratio (e.g., 1:3 ratio). The rapid change in polarity causes lipid self-assembly into LNPs around the RNA strands.
- Buffer Exchange & Purification: Dialyze or use tangential flow filtration (TFF) against a phosphate-buffered saline (PBS) at neutral pH to remove ethanol and buffer components.
- Characterization:
- Size and Polydispersity (PDI): Measure by Dynamic Light Scattering (DLS). Optimal particle size for systemic delivery is 70-100 nm with a PDI < 0.2.
- Encapsulation Efficiency: Quantify using a Ribogreen assay. Compare fluorescence with and without a detergent (e.g., Triton X-100) to distinguish encapsulated from free RNA. >90% encapsulation is typically targeted.
- Zeta Potential: Measure surface charge via electrophoretic light scattering.
In Vitro and In Vivo Functional Assessment
The workflow for testing LNP performance is multi-staged, progressing from cell culture to animal models.
Workflow: Functional Assessment of RNA-LNPs
- In Vitro Transfection:
- Cell Line: Use a relevant cell line (e.g., HepG2 for liver tropism). Seed cells in a 96-well or 24-well plate.
- Dosing: Treat cells with a range of LNP concentrations (e.g., 0.1-100 nM RNA). Include appropriate controls (untreated, empty LNPs).
- Readout:
- For mRNA LNPs: Measure protein expression 24-48 hours post-transfection via fluorescence (if encoding a reporter like GFP) or ELISA (for specific proteins).
- For siRNA LNPs: Quantify mRNA knockdown 48-72 hours post-transfection using qRT-PCR. Assess protein knockdown via Western Blot or flow cytometry.
- In Vivo Efficacy and Biodistribution:
- Animal Model: Administer LNPs to a disease-relevant animal model (e.g., mouse, rat, non-human primate) via the intended clinical route (e.g., intravenous injection).
- Dosing: A common IV dose for siRNA in mice is 1-5 mg RNA per kg body weight.
- Biodistribution: Use LNPs loaded with a reporter mRNA (e.g., Luciferase) for live imaging, or quantify RNA concentration in harvested tissues using qPCR.
- Efficacy: Collect blood or tissue samples at multiple time points to measure pharmacodynamic effects (e.g., target protein knockdown for siRNA, or protein production for mRNA).
The following diagram maps this comprehensive experimental workflow.
Diagram 2: LNP Performance Assessment Workflow. This figure outlines the key stages and associated techniques for the formulation, characterization, and functional evaluation of RNA-loaded lipid nanoparticles.
The Scientist's Toolkit: Essential Reagents and Solutions
Successful development and testing of LNP-based therapies rely on a suite of specialized reagents and tools. The following table details key solutions for researchers in this field.
Table 3: Essential Research Reagent Solutions for LNP Development
| Reagent / Material | Function | Example Use Case |
|---|---|---|
| Ionizable Lipids (e.g., DLin-MC3-DMA, SM-102) | Core functional lipid for RNA complexation and endosomal escape. | Optimizing LNP formulations for potency and reduced immunogenicity [50]. |
| Helper Lipids (DOPE, DSPC) | Modulate LNP fusogenicity and stability. | Comparing DOPE (fusogenic) vs. DSPC (stable) for different tissue targets [50]. |
| PEG-Lipids (DMG-PEG, DSG-PEG) | Stabilize LNPs, control size, and reduce nonspecific uptake. | Screening PEG-lipid chain length and concentration to optimize pharmacokinetics [50]. |
| Microfluidic Devices | Enable reproducible, scalable LNP formation via rapid mixing. | Formulating LNPs with consistent size and high encapsulation efficiency [51]. |
| Ribogreen Assay Kit | Fluorescent quantification of RNA encapsulation efficiency in LNPs. | Determining the percentage of RNA successfully encapsulated versus free RNA after formulation [50]. |
| In Vivo Imaging System (IVIS) | Non-invasive tracking of biodistribution using reporter genes (e.g., Luciferase). | Visualizing and quantifying the organ-level distribution of mRNA-LNPs encoding luciferase in live animals [49]. |
| Parp1-IN-18 | Parp1-IN-18 | Potent PARP1 Research Inhibitor | Parp1-IN-18 is a high-quality PARP1 inhibitor for cancer and DNA repair research. This product is for Research Use Only and not for human or veterinary diagnosis or therapeutic use. |
| Crocapeptin C | Crocapeptin C | Crocapeptin C is a potent, research-grade cyclic depsipeptide inhibiting chymotrypsin (IC50 0.5 µM) and platelet aggregation. For Research Use Only. Not for human use. |
The clinical success of LNP-delivered siRNA and mRNA has unequivocally validated non-viral vectors as a cornerstone of modern gene therapy. However, the current landscape is dominated by applications targeting the liver, highlighting a primary limitation and the focus of ongoing research [49].
The Next Frontier: Extrahepatic Delivery
The future of LNP technology lies in engineering vectors that can efficiently and safely target tissues beyond the liver. Key strategies include:
- Ligand-Targeted LNPs: Conjugating targeting ligands (e.g., antibodies, peptides, sugars like GalNAc) to the LNP surface to achieve active targeting to specific cell types in organs like the lungs, brain, and heart [48] [49]. While GalNAc is well-established for hepatocyte targeting, new ligands are being discovered for other tissues.
- pKa Tuning and Novel Ionizable Lipids: Designing novel ionizable lipids with optimized pKa values to alter charge characteristics and shift biodistribution away from the liver [49].
- Alternative Non-Viral Platforms: Exploring new delivery vehicles such as engineered virus-like particles (eVLPs) for high-efficiency gene editing, polymeric nanoparticles for enhanced stability, and extracellular vesicles (EVs) for their innate biocompatibility and tissue-homing capabilities [48].
Concluding Analysis
In the comparative framework of viral versus non-viral gene delivery, LNPs have carved out a definitive and expanding niche. When benchmarked against viral vectors like AAV and LV, LNPs offer distinct advantages in safety (no risk of genomic integration or pre-existing immunity), manufacturing scalability, and flexibility for transient expression, as required for vaccines or certain protein replacements [3] [25]. The trade-off remains the transient nature of the effect, necessitating repeat administration, which itself is enabled by the lower immunogenicity of LNPs compared to viral vectors.
The experimental data and clinical outcomes summarized in this guide demonstrate that LNP-based RNA delivery is no longer a promising concept but a proven clinical platform. As research in tissue-specific targeting continues to mature, the scope of LNP therapies is poised to expand dramatically, moving beyond the liver to address a wider array of genetic, oncological, and degenerative diseases, thereby solidifying the role of non-viral vectors in the next generation of genetic medicines.
The strategic selection of a drug's administration route is a critical determinant in the success of modern therapeutics, directly governing biodistribution profiles, therapeutic efficacy, and potential side effects. This is particularly paramount in advanced treatment modalities such as gene therapy, radiopharmaceuticals, and nanomedicine, where achieving sufficient drug concentration at the target site while minimizing off-target exposure is a fundamental challenge. The central dichotomy in administration strategy lies between systemic delivery, which circulates a therapeutic agent throughout the entire body via the bloodstream, and localized delivery, which administers the agent directly to or near the target tissue [1] [52].
Understanding the pharmacokinetic and biodistribution consequences of this choice is essential for researchers and drug development professionals designing preclinical and clinical studies. This guide provides a comparative analysis of systemic and localized injection strategies, supported by experimental data and methodologies, to inform decision-making in therapeutic development.
Comparative Analysis of Administration Routes
Quantitative Biodistribution Data
The following tables summarize key quantitative findings from preclinical studies, illustrating how administration routes significantly alter biodistribution and pharmacokinetic parameters.
Table 1: Impact of Administration Route on Tumor and Tissue Uptake
| Therapeutic Agent | Model System | Administration Route | Key Biodistribution Findings | Source |
|---|---|---|---|---|
| 18F-FDG (Radiopharmaceutical Surrogate) | Porcine Orthotopic Renal Tumor | Intra-arterial (IA) + Embolization | 2-4x higher tumor uptake at 1 min (%ID/g: 44.41 ± 2.48) vs. IV; 3x higher concentration at 10 min; trend of lower systemic exposure in blood, liver, kidney, etc. | [53] |
| Intravenous (IV) | Lower initial tumor uptake (%ID/g: 23.19 ± 4.65 at 1 min) and more rapid washout. | |||
| 99mTc-Pamidronate (Bisphosphonate) | Rat Orthopedic Model | Direct Application (d.a.) on bone | 134% higher femoral uptake (%ID/g: 5.15 ± 0.26) at 2h vs. IV; increased to 7.89 ± 0.46 %ID/g with fracture. | [54] |
| Subcutaneous (s.c.) | Significantly lower bone uptake (%ID/g: 0.65 ± 0.07 at 2h). | |||
| Intravenous (i.v.) | Moderate bone uptake (%ID/g: 2.2 ± 0.15 at 2h). | |||
| P12 (Anti-inflammatory Gold Nanoparticle) | LPS-induced Acute Lung Injury (ALI) Mouse | Intratracheal (i.t.) | Superior accumulation in lungs; specific targeting of lung macrophages; better reduction of lung inflammation. | [52] |
| Intravenous (i.v.) | More nanoparticle accumulation in the liver; less in the lungs. | |||
| Intraperitoneal (i.p.) | More accumulation in lymph nodes; less in the lungs. |
Table 2: Pharmacokinetics and Clearance of Nanoparticles and mRNA-LNPs
| Therapeutic Agent | Administration Route | Pharmacokinetic & Clearance Profile | Source |
|---|---|---|---|
| C-dot-ZW800 (Carbon Nanoparticles) | Intravenous (i.v.) | Rapid blood clearance; particle concentration at 1 min was 17.3-fold higher than at 60 min. | [55] |
| Intramuscular (i.m.) | Delayed absorption; particle concentration at 1 min was 9.6-fold lower than at 60 min. | ||
| Subcutaneous (s.c.) | Slowest, sustained absorption; particle concentration at 1 min was 4.4-fold lower than at 60 min. Clearance rate: i.v. > i.m. > s.c. | ||
| mRNA-LNPs (with SM-102 lipid) | Subcutaneous (s.c.) | Superior mRNA protection and ~3x higher bioavailability compared to other lipids (ALC-0315, MC3) after s.c. injection. | [56] |
| Intravenous (i.v.) | Protein expression predominantly in the liver across all formulations. | ||
| Oligonucleotides | Systemic (e.g., i.v.) | Broad biodistribution; highest concentrations in liver and kidney, followed by bone marrow, adipocytes, lymph nodes; does not cross blood-brain barrier. | [57] |
| Local (Intrathecal) | Following intrathecal injection, distributes broadly in the CNS. |
Key Experimental Protocols
To ensure the reproducibility of biodistribution studies, the following details the core methodologies from the cited research.
- Objective: To compare the biodistribution of a small-molecule radiopharmaceutical probe (18F-FDG as a surrogate) after intra-arterial (IA) versus intravenous (IV) delivery, combined with tumor embolization.
- Animal Model: Porcine orthotopic renal tumor model.
- Intervention:
- IA Group: Transcatheter embolization of the tumor's arterioles using micron particles, followed by intra-arterial administration of 18F-FDG directly into the tumor-feeding artery.
- IV Group: Standard intravenous administration of the same dose of 18F-FDG.
- Biodistribution Assessment:
- Animals were imaged at multiple time points (1, 10, and up to 120 minutes post-injection).
- After imaging, animals were euthanized, and target organs (tumor, blood, liver, kidney, spleen, muscle) were harvested.
- Radioactivity in each tissue was measured using a gamma counter and expressed as Percent Injected Dose per Gram of tissue (%ID/g).
- Statistical Analysis: Data were compared between IA and IV groups using appropriate statistical tests (e.g., t-test), with a significance level of P < 0.05.
- Objective: To determine the effect of intratracheal (i.t.), intravenous (i.v.), and intraperitoneal (i.p.) administration on the efficacy and biodistribution of P12 gold nanoparticles in a lung injury model.
- Animal Model: LPS-induced Acute Lung Injury (ALI) mouse model.
- Intervention:
- Mice received P12 nanoparticles conjugated with a Cy5 fluorescent label via one of the three routes (i.t., i.v., or i.p.).
- Biodistribution Assessment:
- Ex Vivo Imaging: At designated time points, major organs (lungs, liver, spleen, kidneys, heart) were harvested and imaged using an in vivo imaging system (IVIS) or similar to quantify fluorescence signal.
- Quantitative Analysis (ICP-MS): The gold core of P12 in tissues was quantified at minuscule levels using Inductively Coupled Plasma Mass Spectrometry (ICP-MS), providing a highly accurate measure of nanoparticle accumulation.
- Cell Targeting: Lung cells were isolated via enzymatic digestion (collagenase II/DNase I), and flow cytometry was performed with antibodies (e.g., against F4/80, CD11b) to identify the cellular populations (e.g., macrophages) that had taken up the P12-Cy5 nanoparticles.
- Therapeutic Efficacy: Lung inflammation and injury were assessed by measuring inflammatory cytokines (e.g., IL-6, MCP-1) in bronchoalveolar lavage fluid (BALF) or lung homogenates via ELISA, and by histological examination of lung sections.
Visualizing Administration Route Impact
The following diagram illustrates the core concepts and findings regarding how administration routes dictate biodistribution and therapeutic outcomes.
The Scientist's Toolkit: Essential Research Reagents
The following table catalogues key reagents and materials used in the featured biodistribution studies, providing a resource for experimental design.
Table 3: Key Research Reagents for Biodistribution Studies
| Reagent / Material | Function / Application | Specific Examples / Notes |
|---|---|---|
| Radiolabels | Quantitative tracking of biodistribution using imaging (PET/SPECT) or gamma counting. | 18F-FDG [53], 99mTc [54]. Allows precise %ID/g measurement. |
| Fluorescent Labels / Dyes | Enables optical imaging (in vivo and ex vivo) and flow cytometry analysis of cell uptake. | Cy5 [52], ZW800 (NIR dye) [55]. Crucial for visualizing distribution and identifying target cells. |
| Animal Disease Models | Provides a physiologically relevant context for evaluating biodistribution and efficacy. | Porcine orthotopic renal tumor [53], LPS-induced ALI mouse [52], various xenograft models. |
| Nanoparticle Systems | Platform for drug/gene delivery; properties (size, charge, surface) dictate in vivo behavior. | Gold Nanoparticles (GNP) [52], Carbon Dots (C-dots) [55], Lipid Nanoparticles (LNP) [56]. |
| Embolization Agents | Used with intra-arterial delivery to block effluent flow, trapping therapeutic agents in the tumor. | Micron particles [53]. Enhances local retention and exposure time. |
| Enzymes for Tissue Processing | Digestion of solid tissues (e.g., lungs, tumors) to create single-cell suspensions for flow cytometry. | Collagenase II, DNase I [52]. |
| Flow Cytometry Antibodies | Identification and characterization of specific cell populations that have internalized a therapeutic. | Antibodies against F4/80, CD11b, CD45, etc. [52]. |
| ELISA Kits | Quantification of therapeutic efficacy via biomarker analysis (e.g., cytokine levels). | Kits for IL-6, MCP-1 [52]. |
| Brd4-IN-5 | Brd4-IN-5, MF:C25H21F2N3O4, MW:465.4 g/mol | Chemical Reagent |
| Antifungal agent 55 | Antifungal agent 55, MF:C18H15BrCl2N2Se, MW:489.1 g/mol | Chemical Reagent |
The choice between systemic and localized administration is a pivotal decision that directly controls the fate of a therapeutic agent in the body. As the data demonstrates, localized routes (such as intra-arterial, intratracheal, or direct application) consistently achieve significantly higher target tissue concentrations and reduce systemic exposure compared to intravenous or other systemic routes. This enhanced local bioavailability can translate to superior therapeutic efficacy and a more favorable safety profile. The optimal strategy is context-dependent, influenced by the target tissue's accessibility, the agent's physicochemical properties, and the disease's pathophysiology. A thorough understanding of these principles is indispensable for designing effective and safe therapeutic interventions.
This guide provides a comparative analysis of four pioneering genetic therapies—Luxturna and Zolgensma (viral vector-based) versus Onpattro and Givlaari (non-viral vector-based)—framed within the broader research context of viral versus non-viral delivery methods for somatic cell gene modification. The analysis covers their respective therapeutic mechanisms, clinical performance, experimental protocols, and practical research considerations to inform scientific and development strategies.
The selected case studies represent two distinct technological approaches: viral vector-mediated gene replacement and non-viral vector-mediated gene silencing.
Table 1: Fundamental Product Characteristics
| Feature | Luxturna | Zolgensma | Onpattro | Givlaari |
|---|---|---|---|---|
| Generic Name | Voretigene neparvovec-rzyl | Onasemnogene abeparvovec-xioi | Patisiran | Givosiran |
| Technology Platform | Viral Vector (AAV2) | Viral Vector (AAV9) | Non-Viral (LNP-siRNA) | Non-Viral (GalNAc-siRNA) |
| Therapeutic Mechanism | Gene Replacement | Gene Replacement | RNA Interference (RNAi) | RNA Interference (RNAi) |
| Target Gene | RPE65 | SMN1 | TTR | ALAS1 |
| Indication | RPE65-mutation associated retinal dystrophy | Spinal Muscular Atrophy (SMA) | hATTR Amyloidosis | Acute Hepatic Porphyria (AHP) |
| Route of Administration | Subretinal injection | Intravenous infusion | Intravenous infusion | Subcutaneous injection |
| Dosing Regimen | One-time administration | One-time administration | Every 3 weeks | Monthly |
Table 2: Clinical and Commercial Performance
| Metric | Luxturna | Zolgensma | Onpattro | Givlaari |
|---|---|---|---|---|
| Key Efficacy Data | Restoration of functional vision measured by MLMT[B:5] | SIGNIFICANT improvement in survival & motor function: 100% of patients in SPR1NT trial sitting independently for ≥30 seconds[B:5] | Significant reduction in neuropathy impairment (mNIS+7) vs. placebo[B:3] | 74% reduction in composite porphyria attack rate in ENVISION trial[B:2] |
| Durability | Long-term expression demonstrated (study follow-up) | Stable transgene expression & sustained clinical outcomes up to 5+ years post-treatment[B:5] | Requires chronic, recurring dosing | Requires chronic, recurring dosing; sustained effect with continued dosing[B:6] |
| Commercial Uptake | ~$250M total sales (2018-2023); treated most eligible patients (est. 650 in US)[B:5] | >$1.2B global sales in 2023; ~80% share in US newly diagnosed SMA patients[B:5] | First RNAi therapy approved; faces competition from other TTR-directed therapies[B:6] | Slow initial uptake; longer-term data show sustained effect[B:6] |
| Price | ~$850,000 per treatment | ~$2.1 million per treatment | ~$450,000 annually | ~$575,000 annually[B:10] |
Therapeutic Mechanisms and Experimental Protocols
Viral Vector-Mediated Gene Replacement (Luxturna, Zolgensma)
This approach utilizes recombinant adeno-associated virus (rAAV) vectors engineered to deliver a functional copy of a defective gene into the nucleus of target cells. The vector does not integrate into the host genome but remains episomal, enabling long-term gene expression in non-dividing or slowly-dividing cells[B:1][B:7].
Diagram 1: rAAV Gene Replacement Mechanism
Key Experimental Workflow for rAAV Clinical Trials:
- Vector Engineering & Packaging: The therapeutic transgene (e.g., RPE65 cDNA for Luxturna, SMN1 cDNA for Zolgensma) is cloned into a plasmid between AAV inverted terminal repeats (ITRs). This ITR-flanked "payload" plasmid is co-transfected with a second "helper" plasmid encoding the AAV rep and cap genes into producer cells (e.g., HEK293) to package the recombinant genome into the viral capsid[B:1].
- Preclinical Animal Studies: Vector potency, tropism, and safety are evaluated in animal models. For Zolgensma, AAV9 was selected for its ability to cross the blood-brain barrier and efficiently transduce motor neurons after systemic administration[B:1][B:5].
- Clinical Delivery: Luxturna is administered via subretinal injection, a localized delivery method requiring specialized surgical protocol to detach the retina and instill the vector directly near retinal pigment epithelial cells[B:3]. Zolgensma is administered via intravenous infusion, a systemic delivery method that requires careful patient management[B:5].
Non-Viral Vector-Mediated Gene Silencing (Onpattro, Givlaari)
This approach employs synthetic delivery systems to transport small interfering RNA (siRNA) to the liver, where they silence the expression of a disease-causing gene by mediating the degradation of its mRNA[B:4].
Diagram 2: Non-Viral RNAi Therapeutic Mechanism
Key Experimental Workflow for RNAi Clinical Trials:
- siRNA Design and Conjugation: The siRNA duplex is designed to be complementary to the target mRNA (e.g., TTR for Onpattro, ALAS1 for Givlaari). For Givlaari and other GalNAc-conjugated therapies, the siRNA is chemically conjugated to a N-acetylgalactosamine (GalNAc) ligand, which targets the asialoglycoprotein receptor (ASGPR) highly expressed on hepatocytes[B:3][B:4].
- Formulation: Onpattro is formulated into Lipid Nanoparticles (LNPs) that protect the siRNA from degradation and facilitate its delivery into the cytoplasm of hepatocytes after intravenous administration[B:3][B:7].
- Clinical Dosing: Unlike one-time viral therapies, RNAi therapies like Onpattro and Givlaari require chronic administration (e.g., every 3 weeks for Onpattro, monthly for Givlaari) to maintain therapeutic effect, as they do not permanently alter gene expression but instead continuously degrade newly produced mRNA[B:2][B:6].
Comparative Analysis: Viral vs. Non-Viral Delivery
Table 3: Strategic Comparison of Delivery Platforms
| Aspect | Viral Vectors (AAV) | Non-Viral Vectors (LNP/GalNAc) |
|---|---|---|
| Mechanism | Gene Replacement: Delivers a correct gene copy to produce a functional protein. | Gene Silencing: Uses siRNA to degrade mRNA, reducing levels of a pathogenic protein. |
| Therapeutic Profile | Potential for one-time, curative treatment with long-lasting expression. | Typically requires chronic, recurring dosing to maintain effect. |
| Key Advantage | High transduction efficiency and durable expression in non-dividing cells. | Favorable safety profile (low immunogenicity, no insertional mutagenesis risk)[B:7]. |
| Key Challenge | Immunogenicity: Pre-existing or treatment-induced immune responses can limit efficacy[B:1][B:7]. Manufacturing complexity and high production costs[B:7][B:9]. | Transfection Efficiency: Historically lower than viral methods, though advanced formulations (LNP, GalNAc) have improved this significantly[B:7]. |
| Cargo Capacity | Limited (<~5 kb), constraining the size of the deliverable transgene[B:7]. | Larger cargo capacity possible, more flexible for payload design[B:7]. |
| Manufacturing & Scalability | Complex, expensive, difficult to scale due to biological production systems[B:7][B:9]. | Generally simpler, more scalable, and lower-cost chemical synthesis[B:7]. |
The Scientist's Toolkit: Key Research Reagents
Table 4: Essential Research Materials for SMGT Development
| Reagent / Material | Function in Research | Example Application in Featured Cases |
|---|---|---|
| Plasmid DNA System (ITR-flanked) | Contains the therapeutic gene expression cassette flanked by AAV Inverted Terminal Repeats (ITRs), which are the only viral components required for replication and packaging[B:1]. | Used in the initial cloning and production of rAAV vectors like those for Luxturna and Zolgensma. |
| Packaging Cell Line (e.g., HEK293) | A cell line used to produce recombinant viral vectors. It often supplies essential viral genes (e.g., Adenovirus E1) in trans to facilitate AAV replication[B:1]. | Standard industry platform for manufacturing clinical-grade AAV vectors. |
| AAV Serotype Capsids (e.g., AAV2, AAV9) | The protein shell of the virus determining tropism (which cells/organs the vector can infect). Different serotypes are selected for specific tissue targeting[B:1]. | AAV2 for retinal transduction (Luxturna); AAV9 for crossing the blood-brain barrier (Zolgensma). |
| Cationic Lipids / LNPs | Positively charged lipids that form complexes with nucleic acids, protecting them and promoting cellular uptake and endosomal escape[B:7]. | Critical delivery vehicle for Onpattro (patisiran), enabling siRNA delivery to hepatocytes. |
| GalNAc Ligand | A carbohydrate ligand that specifically binds to the ASGPR receptor on hepatocytes, enabling highly targeted delivery of conjugated therapeutics[B:3][B:4]. | Conjugated to Givlaari (givosiran) siRNA for targeted liver delivery without the need for complex formulations like LNPs. |
| siRNA Duplex | A synthetic RNA molecule designed to be complementary to a target mRNA sequence, which upon delivery into the cell, directs the RNA-induced silencing complex (RISC) to cleave the target mRNA[B:4]. | The active pharmaceutical ingredient in both Onpattro and Givlaari. |
Research and Development Implications
The choice between viral and non-viral delivery systems is fundamental and depends on the therapeutic objective, target tissue, and desired duration of effect.
- Opt for Viral Vectors (AAV) when: The goal is long-term or permanent correction of a genetic defect via gene replacement or addition, particularly for monogenic disorders affecting non-regenerating tissues (e.g., retina, CNS, muscle). The high transduction efficiency and durable expression justify the development complexity and immunogenicity challenges[B:1][B:7].
- Opt for Non-Viral Vectors (LNP/GalNAc) when: The goal is to reduce the expression of a disease-causing gene, especially for liver-associated diseases. This approach is advantageous due to its simpler manufacturing, superior safety profile, and adaptability for chronic administration where durable effect is not possible or required[B:3][B:4][B:7].
Future directions in somatic cell gene therapy research will focus on overcoming the limitations of both platforms, including engineering novel AAV capsids with enhanced tropism and reduced immunogenicity, developing non-viral systems capable of efficient in vivo delivery beyond the liver, and advancing gene editing technologies that may combine the best attributes of both viral and non-viral delivery systems.
Overcoming Technical Hurdles: Safety, Manufacturing, and Efficiency Challenges
Viral vectors have emerged as a leading platform for gene delivery, demonstrating remarkable success in treating both inherited and acquired diseases [14] [58]. Their ability to efficiently transduce target cells and provide sustained therapeutic gene expression has resulted in several approved medicines, including Luxturna for inherited retinal disease and Zolgensma for spinal muscular atrophy [58]. However, host immune responses to these vectors constitute one of the most significant barriers to their widespread clinical application [58] [59]. The mammalian immune system has evolved sophisticated mechanisms to recognize and eliminate viral invaders, and these same mechanisms perceive viral vectors as potential threats despite their therapeutic intent [58]. These immune responses can manifest as both innate reactions occurring within hours of administration and adaptive responses developing over days to weeks, ultimately reducing transduction efficiency, eliminating transduced cells, and limiting the duration of therapeutic expression [58] [59]. Furthermore, pre-existing immunity to viral vectors from natural infections can completely neutralize therapeutic efficacy upon first administration [58] [60]. This comprehensive review examines the immune challenges associated with leading viral vector platforms and systematically compares the strategies being developed to overcome these critical barriers.
Comparative Immunogenicity of Viral Vector Platforms
The immunogenic profile varies considerably among different viral vector platforms, influencing their suitability for specific therapeutic applications. The table below summarizes key immune characteristics of the three most widely used viral vector systems.
Table 1: Comparative Immune Profiles of Major Viral Vector Platforms
| Vector Platform | Innate Immune Activation | Pre-existing Immunity in Humans | Adaptive Immune Responses | Primary Immune Concerns |
|---|---|---|---|---|
| Adenovirus (Ad) | Potent; triggers inflammation, thrombocytopenia, endothelial activation [58] [59] | High to common human serotypes [58] | Strong CD8+ T cell responses to viral genes; neutralizing antibodies [58] [59] | Immunotoxicity; elimination of transduced cells; neutralization [58] |
| Adeno-Associated Virus (AAV) | Comparatively weak and transient; TLR9 signaling; complement activation [58] [59] | Varies by serotype and geography [58] [59] | CD8+ T cells to capsid; neutralizing antibodies; generally less efficient at inducing CD8+ T cells to transgene [58] [59] | Capsid-specific T cells destroying transduced cells; antibody-mediated neutralization [59] [60] |
| Lentivirus (LV) | Strong IFN-α/β response; DC activation [58] [59] | Low [58] | Efficient T and B cell responses to transgene unless regulated; possible responses to envelope [58] [59] | Responses to transgene product; potential insertional mutagenesis [24] |
Adenoviral Vectors: Balancing Potency and Reactogenicity
Adenoviral vectors are among the most immunogenic viral vector platforms, triggering robust innate and adaptive immune responses [58]. Systemically delivered Ad vectors activate a broad spectrum of innate immune pathways, resulting in rapid production of inflammatory cytokines, activation of vascular endothelial cells, and platelet aggregation that can lead to thrombocytopenia [58] [59]. These early responses create an inflammatory environment that primes adaptive immunity, leading to strong CD8+ T cell responses against both viral antigens and the therapeutic transgene product [58]. The Ad vector's efficiency in transducing antigen-presenting cells (APCs) further enhances its immunogenicity, making it particularly effective for vaccine applications but problematic for sustained gene replacement therapies [58]. Consequently, research has increasingly focused on their use in cancer gene therapy and as vaccine carriers rather than for traditional gene replacement [58].
Adeno-Associated Viral Vectors: The Challenge of Capsid-Specific Immunity
AAV vectors are generally considered less immunogenic than adenoviral vectors but face significant challenges related to both pre-existing and treatment-induced immunity [58] [59]. While AAV triggers relatively weak and transient innate immune responses compared to Ad vectors, it does activate Toll-like receptor 9 (TLR9) signaling and the complement system, which can promote CD8+ T cell responses [59]. The primary immunological hurdle for AAV therapies involves capsid-specific adaptive immune responses [60]. Pre-existing neutralizing antibodies (NABs) to various AAV serotypes are common in human populations and can completely prevent successful transduction upon vector administration [59]. Perhaps more concerning is the development of capsid-specific CD8+ T cells following AAV administration, which can eliminate transduced hepatocytes and other target cells, leading to loss of therapeutic expression [59] [60]. This phenomenon was clearly demonstrated in a clinical trial for hemophilia B, where a patient receiving hepatic AAV gene transfer experienced a decline in factor IX expression accompanied by elevated liver enzymes and the emergence of AAV capsid-specific T cells in peripheral blood [59].
Lentiviral Vectors: Managing Responses to Transgene Products
Lentiviral vectors trigger strong type I interferon (IFN-α/β) responses that can limit transduction efficiency and drive subsequent adaptive immunity [58] [59]. Unlike AAV vectors, LV vectors efficiently transduce professional antigen-presenting cells, particularly dendritic cells, which contributes to their ability to generate robust immune responses to the transgene product [58] [59]. This immunogenicity has been strategically harnessed for ex vivo modification of T cells in CAR-T therapies but presents challenges for in vivo gene therapy applications where sustained expression is desired [58] [24]. A key safety consideration with earlier generations of retroviral vectors, including LVs, was the risk of insertional mutagenesis due to semi-random integration into the host genome, though self-inactivating (SIN) configurations have significantly improved their safety profile [24].
Advanced Strategies to Circumvent Vector Immunogenicity
Engineering Immune-Stealth Vectors
Novel bioengineering approaches are creating viral vectors with reduced immunogenicity. For AAV vectors, strategies include site-directed mutagenesis of surface-exposed tyrosine residues to reduce antigen presentation and the development of capsid shuffling techniques to create novel AAV variants resistant to neutralization by human sera [59]. The most innovative approach involves the creation of biomimetic artificial enveloped viral (AEV) vectors designed to shield AAVs from immune recognition [61]. These AEV vectors incorporate AAV cores within synthetic lipid membranes that mimic natural enveloped viruses, effectively protecting them from neutralizing antibodies and reducing uptake by antigen-presenting cells [61]. In murine studies, AEVs demonstrated superior transduction efficiency compared to conventional AAVs in both primary and secondary injections, even in the presence of pre-existing immunity, highlighting their potential for re-administration [61].
Table 2: Experimentally Validated Strategies for Mitigating Immune Responses to Viral Vectors
| Strategy Category | Specific Approach | Experimental Model | Key Outcome | Reference |
|---|---|---|---|---|
| Capsid/Envelope Engineering | Artificial Enveloped Viruses (AEV) | Mouse models of hemophilia B and lymphoma | Enabled re-administration despite pre-existing antibodies; reduced immune activation | [61] |
| Capsid Engineering | Tyrosine residue modification | In vitro antigen presentation assays | Reduced MHC I presentation and CD8+ T cell activation | [59] |
| Capsid Engineering | Serotype switching (AAVrh32.33) | Human serum neutralization assays | Reduced neutralization by human sera | [59] |
| Immunosuppression | Unknown agent | Clinical trial for hemophilia B | Prevention of T-cell mediated loss of transduced hepatocytes | [59] |
| Vector Genome Design | miRNA-regulated expression in APCs | Lentiviral vectors in mouse models | Reduced immune responses to transgene product | [59] |
Immunomodulatory Protocols
Pharmacological immunosuppression represents a complementary approach to manage immune responses against viral vectors [59]. While the exact protocols vary, transient immunosuppression around the time of vector administration has shown promise in preventing T-cell mediated elimination of transduced cells in clinical trials [59]. This approach was investigated following observations in a hemophilia B trial where a patient developed capsid-specific T cells that correlated with declining factor IX expression [59]. The timing and duration of immunosuppression are critical factors, with protocols typically initiating before or concurrently with vector administration to effectively modulate both the initial innate response and subsequent adaptive immunity [59].
Route-Specific Immune Outcomes
The administration route significantly influences the immunogenicity of viral vectors, with some delivery sites demonstrating increased propensity for immune tolerance induction [59]. Hepatic gene transfer has emerged as a particularly favorable route, associated with induction of regulatory T cells (Tregs) and immune tolerance to the transgene product [59]. This phenomenon is attributed to the liver's unique immunomodulatory environment and the presence of specialized antigen-presenting cells that promote tolerance rather than activation [59]. In contrast, intramuscular delivery may elicit stronger immune responses to the transgene product, though it benefits from relative isolation from circulating neutralizing antibodies [59]. These route-dependent immunological outcomes enable strategic selection of administration sites based on the specific therapeutic goals—favoring intramuscular delivery for vaccines where robust immune activation is desired, and hepatic delivery for protein replacement therapies where sustained expression without immune elimination is critical.
Experimental Approaches for Evaluating Vector Immunogenicity
Standardized Assays for Immune Monitoring
Comprehensive assessment of immune responses to viral vectors requires a multifaceted experimental approach. Key methodologies include:
- Enzyme-Linked Immunosorbent Assay (ELISA) for quantifying vector-specific antibody titers, particularly neutralizing antibodies that prevent cellular transduction [59] [60].
- Enzyme-Linked Immunospot (ELISpot) assay for detecting antigen-specific T cells through interferon-γ production, crucial for identifying capsid-reactive cellular immunity [59].
- Flow cytometric analysis of intracellular cytokines and surface activation markers to characterize functional T cell responses and memory formation [59] [60].
- Tetramer staining for direct quantification of capsid-specific CD8+ T cells, providing high specificity for tracking cytotoxic T lymphocyte populations [59].
These assays are typically performed sequentially following vector administration in preclinical models and clinical trials, with baseline measurements followed by longitudinal monitoring to capture the evolution of immune responses over time [59] [60].
In Vivo Models for Immunogenicity Assessment
Animal models remain indispensable for evaluating vector immunogenicity, though significant species-specific differences exist [59]. Murine models provide accessible systems for initial screening of immune responses but often fail to fully recapitulate human immunity, particularly for AAV vectors where capsid-specific T cell responses observed in humans have been difficult to replicate in mice [59]. Non-human primates offer closer immunological similarity to humans and are valuable for assessing the impact of pre-existing immunity, but they come with substantial ethical and cost considerations [59]. The development of humanized mouse models containing elements of the human immune system represents a promising approach to bridge this translational gap, allowing investigation of human-relevant immune responses in a more accessible platform [59].
The following diagram illustrates the key innate immune sensing pathways for viral vectors and the experimental workflow for evaluating immunogenicity:
Diagram 1: Immune Recognition Pathways and Experimental Assessment of Viral Vector Immunogenicity. This diagram illustrates the key innate immune sensing mechanisms for viral vectors (left) and the experimental methods for monitoring immune responses (right). Abbreviations: PRR (Pattern Recognition Receptors), TLR9 (Toll-like Receptor 9), DAMPs (Damage-Associated Molecular Patterns), NAb (Neutralizing Antibodies).
The Scientist's Toolkit: Essential Reagents for Immunogenicity Studies
Table 3: Key Research Reagents for Investigating Viral Vector Immunogenicity
| Reagent/Cell System | Primary Application | Experimental Function |
|---|---|---|
| Human PBMCs | In vitro immunogenicity screening | Assess T cell responses to capsid and transgene antigens |
| TLR9 knockout mice | Innate immunity studies | Define role of nucleic acid sensing in AAV immunogenicity |
| Complement factor-deficient serum | Complement pathway analysis | Determine contribution of complement to vector clearance |
| HLA-transgenic mice | Human-relevant T cell epitope mapping | Identify immunodominant capsid epitopes restricted by human MHC |
| Recombinant AAV capsid proteins | T cell assays | Stimulate capsid-specific T cells for functional characterization |
| Neutralizing antibody standards | Serology assays | Quantify and standardize NAb measurements across studies |
| Cytokine multiplex arrays | Immune monitoring | Profile inflammatory responses to vector administration |
The immunogenicity of viral vectors remains a formidable challenge that requires sophisticated, multi-pronged approaches tailored to specific vector platforms and therapeutic applications. While adenoviral vectors offer high transduction efficiency, their potent immunogenicity must be carefully managed through vector engineering or harnessed for immunostimulatory applications like vaccines and cancer therapy. AAV vectors, despite their favorable safety profile, face significant hurdles related to pre-existing immunity and capsid-specific T cell responses that are being addressed through novel capsid engineering and immune-stealth technologies like artificial enveloped viruses. Lentiviral vectors present distinct challenges related to transgene-specific immunity, particularly for in vivo applications, though these can be mitigated through careful vector design incorporating regulatory elements that limit expression in antigen-presenting cells. The optimal strategy for mitigating immune responses will likely combine multiple approaches—including vector engineering, route optimization, and transient immunomodulation—to achieve the delicate balance between therapeutic efficacy and acceptable immune recognition. As these technologies mature, they will expand the therapeutic potential of viral vector gene therapies to broader patient populations and a wider range of genetic disorders.
Insertional mutagenesis is a potentially serious adverse event in gene therapy, occurring when the integration of a therapeutic gene vector disrupts or alters the function of a host cell's genes. This phenomenon is a primary safety concern for integrating vector systems, as it can lead to the activation of proto-oncogenes or inactivation of tumor suppressor genes, potentially resulting in clonal expansion and malignancies [62] [63]. The risk was starkly illustrated in early clinical trials for X-linked Severe Combined Immunodeficiency (SCID-X1), where use of gamma-retroviral vectors led to T-cell leukemia in several patients due to insertional activation of the LMO2 proto-oncogene [62] [64].
The genotoxic risk profile varies significantly between gene delivery systems, influencing both preclinical safety assessments and clinical trial design. This guide provides a comparative analysis of insertional mutagenesis risks across viral and non-viral gene therapy platforms, supported by experimental data and safety records from clinical applications. Understanding these risks and the development of mitigation strategies is crucial for researchers and drug development professionals advancing gene therapies toward clinical use [62] [64].
Mechanisms of Insertional Mutagenesis
The genotoxicity of integrating vectors manifests through several molecular mechanisms, with the outcome dependent on both the vector design and the biological context of the integration site.
Primary Molecular Mechanisms
- Enhancer-Mediated Activation: Vector-integrated enhancer elements can activate neighboring proto-oncogenes from a significant distance. This was the dominant mechanism observed in the SCID-X1 trial leukemias, where retroviral enhancers activated LMO2 expression [62] [63].
- Promoter Insertion: Integration near the start site of a proto-oncogene can bring it under the control of a strong viral promoter, leading to aberrant expression [64].
- Gene Disruption: Insertion within a tumor suppressor gene can disrupt its coding sequence or lead to truncated, non-functional proteins [64].
- Altered Splicing: Integration within intronic regions can disrupt normal RNA splicing patterns, potentially generating oncogenic fusion proteins or inactivating tumor suppressors [64].
The following diagram illustrates these core mechanisms and their potential consequences leading to clonal expansion.
Influencing Factors on Genotoxic Outcome
The probability that a vector integration event leads to a malignant transformation is not uniform and depends on several biological factors:
- Target Cell Type: Hematopoietic stem cells (HSCs) with self-renewal capacity present higher long-term risk compared to terminally differentiated cells [64]. Clinical experience shows that while HSC gene therapy resulted in leukemias, viral-mediated transduction of T-cells for adoptive immunotherapy has not produced similar adverse outcomes [64].
- Disease Context: The underlying disease can influence risk. In SCID-X1, the γC cytokine receptor transgene may have provided a proliferative advantage that synergized with LMO2 activation [62]. In contrast, ADA-SCID trials using similar vectors showed no leukemias despite insertions near proto-oncogenes like LMO2 [62].
- Proliferative Pressure: Conditions that drive extensive proliferation of corrected cells may increase the likelihood that a rare mutagenic event will expand clonally. The therapeutic transgene itself can provide such pressure if it participates in growth signaling pathways [62].
Comparative Genotoxicity of Delivery Systems
Different gene delivery systems exhibit distinct integration profiles and genotoxic risk based on their biological origins and engineered characteristics.
Viral Vector Systems
Table 1: Comparative Genotoxicity Profiles of Viral Vector Systems
| Vector Type | Integration Profile | Oncogene Activation Cases | Key Safety Features | Clinical Status |
|---|---|---|---|---|
| Gamma-Retroviral Vectors | Preferential integration near transcription start sites and regulatory regions [64] | 5/20 SCID-X1 patients (LMO2, CCND2) [62]; 2/2 X-CGD patients (MDS1/Evi1) [62] | First-generation vectors with intact LTRs; being superseded by SIN designs [26] | No longer selected for new clinical trials [25] |
| Lentiviral Vectors | Prefers integration within active transcriptional units [26] | No reported malignancies in clinical trials to date [64] | Self-inactivating (SIN) designs with deleted viral promoters/enhancers [26] | Becoming standard for ex vivo therapy (e.g., Zynteglo, Libmeldy) [65] [3] |
| Adeno-Associated Virus (AAV) | Predominantly episomal; rare genomic integration [25] | No direct oncogene activation cases reported | Limited integration capacity; primarily remains episomal [25] | Multiple approved products (Luxturna, Zolgensma) [3] |
| Adenovirus | Non-integrating; remains episomal [66] | No insertional mutagenesis risk | Non-integrating nature eliminates insertional risk [66] | Used in vaccines and oncology (GENDICINE) [3] |
Non-Viral Vector Systems
Table 2: Genotoxicity Profiles of Non-Viral Gene Delivery Systems
| Vector Type | Integration Mechanism | Genotoxic Risk | Key Safety Features | Clinical Status |
|---|---|---|---|---|
| Sleeping Beauty Transposon | Cut-and-paste transposition; minimal site preference [64] | Theoretical risk; no clinical cases reported | No viral sequences; minimal site preference compared to retroviruses [64] | In clinical trials for CAR-T applications [64] |
| Lipid Nanoparticles (LNPs) | Primarily for mRNA delivery; non-integrating [67] | No insertional mutagenesis risk | Transient expression; no genomic integration [67] [3] | Approved for siRNA (Onpattro) and vaccines [3] |
| GalNAc Conjugates | RNA-based; non-integrating [3] | No insertional mutagenesis risk | Tissue-targeted (liver); transient effect [3] | Multiple approved products (Givlaari, Oxlumo) [3] |
| mRNA Platforms | Non-integrating; cytoplasmic expression [67] | No insertional mutagenesis risk | No nuclear entry; transient duration [67] | Approved vaccines; therapeutic applications in development |
Clinical Safety Records by Disease Context
Table 3: Clinical Adverse Events in Hematopoietic Stem Cell Gene Therapy Trials
| Disease | Patients Treated | Vector Type | Adverse Events (Leukemia/MDS) | Insertion Sites (Oncogenes) | Clinical Outcome |
|---|---|---|---|---|---|
| SCID-X1 | 9 (French trial) [62] | γ-retroviral (MFG) | 4/9 T-ALL [62] | LMO2 (3/4), CCND2 (1/4), BMI1 (1/4) [62] | 1 death; 3 remission with chemotherapy [62] |
| SCID-X1 | 10 (UK trial) [62] | γ-retroviral (MFG) | 1/10 T-ALL [62] | LMO2 [62] | Remission with chemotherapy [62] |
| ADA-SCID | 10 [62] | MLV-based retroviral | None [62] | Insertions near DYRK1A, BLM, LMO2, CCND2, BCL2; no clonal selection [62] | All alive with improved immune function [62] |
| X-CGD | 2 [62] | γ-retroviral (pSF7, SFFV) | 2/2 MDS [62] | MDS1/Evi1, PRDM16, SETBP1 [62] | 1 death from sepsis; 1 underwent transplant [62] |
| X-ALD | 2 [62] | HIV-1-derived lentiviral | None [62] | No clonal dominance [62] | Alive with decreased disease progression [62] |
| WAS | 2 [62] | Retroviral (CMMP) | 1/2 T-ALL [62] | LMO2, CCND2, BMI1 in T-cells [62] | 1 with ongoing chemotherapy; 1 alive with improved function [62] |
Experimental Assessment of Genotoxicity
Robust preclinical assessment is critical for evaluating the genotoxic potential of novel gene therapy vectors. The following experimental approaches represent standard methodologies in the field.
Insertion Site Analysis
Purpose: To characterize the genomic distribution of vector integrations and identify clusters near cancer-associated genes.
Protocol:
- Genomic DNA Extraction: High-quality DNA from transduced cells (≥5×10ⶠcells recommended) [62]
- Integration Site Amplification: Linear amplification-mediated PCR (LAM-PCR) or non-restrictive LAM-PCR for genome-wide coverage [62]
- High-Throughput Sequencing: Illumina sequencing to generate millions of integration site sequences
- Bioinformatic Analysis: Mapping to reference genome (e.g., hg38) with identification of genes within ±50kb of integration sites
- Statistical Analysis: Compare observed vs. expected integration frequency near oncogenes using Fisher's exact test; identify common integration sites in expanded clones [62]
Interpretation: Clonal expansion monitored over time provides the most relevant risk assessment, as most oncogene-proximal integrations do not cause transformation [64].
In Vitro Transformation Assays
Purpose: To evaluate the direct transforming potential of vector systems in sensitive cell models.
Protocol:
- Cell Model Preparation: Immortalized murine hematopoietic progenitor cells (e.g., BM185) or primary human CD34⺠cells [62]
- Vector Transduction: Optimized MOI to achieve 30-50% transduction without excessive multiplicity
- Clonogenic Assay: Culture in methylcellulose with relevant cytokines; count colonies after 7-14 days
- Serial Replating: Repeated plating of colonies to assess self-renewal capacity beyond normal limits
- Dominant Clone Analysis: Sequence integration sites from predominant colonies for oncogene association [62]
Validation: The in vitro transformation frequency should correlate with known in vivo genotoxicity profiles of reference vectors.
In Vivo Tumorigenesis Models
Purpose: To assess genotoxic risk in physiological context with immune surveillance and tissue microenvironments.
Protocol:
- Mouse Strain Selection: Immunodeficient NSG mice for human cell engraftment or syngeneic models for murine cells
- Cell Transplantation: Transduced hematopoietic stem cells (100-500k CD34⺠cells) via tail vein injection after conditioning
- Long-Term Monitoring: Peripheral blood sampling every 4 weeks for vector copy number and clonal diversity assessment
- Terminal Analysis: Necropsy at 6-12 months for histopathology; integration site analysis in dominant clones [62]
- Control Groups: Include mock-transduced and positive control (e.g., first-generation γ-retroviral vector) groups
The following diagram illustrates the complete workflow for preclinical genotoxicity assessment, from initial vector testing to final analysis.
Research Reagent Solutions
Table 4: Essential Research Reagents for Genotoxicity Assessment
| Reagent/Category | Specific Examples | Research Application | Key Features |
|---|---|---|---|
| Vector Systems | SIN Lentiviral vectors [26], Sleeping Beauty transposon [64] | Comparative genotoxicity studies | Reduced enhancer activity; improved safety profiles |
| Cell Separation | CD34⺠isolation kits [62] | HSC transduction studies | High-purity hematopoietic stem cells for transduction |
| Cell Culture | Serum-free expansion media [62], Cytokine cocktails | In vitro transformation assays | Supports progenitor growth without differentiation |
| Integration Site Mapping | LAM-PCR kits [62], Next-generation sequencing | Genomic integration analysis | Genome-wide coverage; high sensitivity for rare clones |
| Animal Models | Immunodeficient mice (NSG) [62] | In vivo tumorigenesis studies | Supports human hematopoiesis; enables long-term monitoring |
| Bioinformatics | Integration site analysis pipelines [62] | RIS analysis | Identifies common integration sites; statistical analysis |
The management of genotoxic risk in gene therapy requires careful consideration of vector selection, disease context, and target cell biology. Viral vectors, particularly gamma-retroviruses, demonstrate significant genotoxicity in clinical applications, especially in SCID-X1 and CGD trials. Lentiviral vectors with self-inactivating designs show improved safety profiles, while AAV vectors present minimal insertional risk due to predominantly episomal persistence. Non-viral systems including transposons and RNA/LNP platforms offer promising alternatives with potentially safer profiles, though long-term clinical data is still emerging.
Robust preclinical assessment using the described experimental protocols remains essential for predicting and mitigating genotoxic risk. As the field advances, the combination of safer vector designs, improved transduction methods, and enhanced surveillance protocols will enable broader application of gene therapies while minimizing the risk of insertional mutagenesis.
The successful clinical application of gene therapies is profoundly dependent on the scalable and robust manufacturing of their delivery vectors. Both viral and non-viral vector systems present distinct and significant challenges in scaling production from laboratory bench to commercial scale, impacting the cost, consistency, and global availability of these transformative treatments [68] [69]. Viral vectors, including lentivirus (LV) and adeno-associated virus (AAV), are renowned for their high transduction efficiency but are hampered by complex biology and costly production processes [24] [70]. Non-viral methods, such as lipid nanoparticles (LNPs) and electroporation, offer advantages in safety and scalability but have historically faced hurdles in achieving high transfection efficiency and overcoming off-target biodistribution, often leading to accumulation in the liver [15] [3]. This guide provides an objective comparison of these platforms, focusing on their manufacturing complexities, and presents key experimental data and protocols essential for process development scientists and drug development professionals.
Comparative Analysis of Manufacturing Complexities
The journey from research to commercial product involves navigating critical manufacturing hurdles. The table below summarizes the primary challenges and current solutions for both viral and non-viral vector production systems.
Table 1: Key Manufacturing Challenges and Scaling-Up Strategies for Gene Therapy Vectors
| Manufacturing Aspect | Viral Vector Challenges | Viral Vector Scaling Strategies | Non-Viral Vector Challenges | Non-Viral Vector Scaling Strategies |
|---|---|---|---|---|
| Production Platform | Reliance on transient transfection of adherent cells; producer cell toxicity [70]. | Shift to suspension cell cultures in bioreactors; development of inducible producer cell lines [70]. | Optimization of nanoparticle formulations for stability and efficiency [15]. | Use of established scalable methods like microfluidics for LNP formation; design of novel polymers [9] [15]. |
| Process Cost & Scalability | High cost of goods; lentiviral vector production can account for ~40% of total cost for CAR-T therapies [70]. | Holistic system optimization (cell line, media, reagents) to improve titers and reduce volume requirements [70]. | Lower production costs relative to viral methods, but challenges in achieving tissue-specific targeting [3]. | Conjugation with targeting ligands (e.g., GalNAc for liver) to improve specificity; exploration of novel vehicles like proteolipid vehicles (PLVs) [9] [3]. |
| Product Characterization & Consistency | Complex product characterization; challenges with analytical method throughput, resolution, and reproducibility [69]. | Implementation of Quality by Design (QbD); use of stress studies and ultra-scale down tools for process acceleration [69]. | Controlling nanoparticle size, charge, and stability to avoid aggregation and ensure consistent performance [15]. | Rigorous optimization of parameters like polymer hydrophobicity, PEG density, and charge density to ensure batch-to-batch consistency [15]. |
| Immunogenicity & Safety | Risk of immune responses and insertional mutagenesis [24] [3]. | Engineering of self-inactivating (SIN) vectors; use of tissue-specific promoters and capsid engineering [24] [3]. | Generally lower immunogenicity, but potential cytotoxicity with cationic materials [15]. | Development of highly hydrophilic and biodegradable materials to reduce cytotoxicity and protein adsorption [15]. |
Quantitative Manufacturing Data Comparison
To further illustrate the scaling landscape, the following table consolidates key quantitative data related to the production and performance of these systems.
Table 2: Quantitative Data Comparison for Viral and Non-Viral Vector Production
| Parameter | Lentiviral Vectors | AAV Vectors | Lipid Nanoparticles (LNPs) |
|---|---|---|---|
| Cargo Capacity | ~10 kb [24] | ~4.7 kb [3] | Versatile (DNA, mRNA, siRNA, CRISPR) [9] |
| Production Scale | Moving from flasks to >200 L bioreactors [70] | Facing demands for 1-2 order magnitude increase in capacity [68] | Established large-scale production for vaccines [9] |
| Production Cost | Up to 40% of total COGs for CAR-T therapies [70] | High, due to complex biology and analytics [69] | Lower cost, ease of synthesis [15] |
| Critical Quality Attributes | Titer, infectivity, absence of replication-competent lentiviruses | Full/empty capsid ratio, potency, aggregation [69] | Size, polydispersity, encapsulation efficiency, pKa [15] |
Experimental Protocols for Process Development
Protocol: Optimizing Lentiviral Vector Production in Suspension Culture
This protocol is adapted from efforts to develop a cost-effective, high-titer lentiviral manufacturing process with a reduced labor burden for scale-up in stirred-tank reactors (STRs) [70].
- Step 1: Cell Line and Media Selection: Utilize a serum-free suspension cell line (e.g., HEK 293-derived) adapted to chemically defined medium. The selection of a robust, high-density cell line is foundational.
- Step 2: Transfection Optimization: Perform a design of experiment (DoE) to optimize transfection conditions. Critical parameters include:
- Cell Density at Transfection: Typically between 1-4 x 10^6 cells/mL.
- DNA Quantity and Ratio: Optimize the total amount of plasmid DNA and the ratio of transfer, packaging, and envelope plasmids.
- Transfection Reagent: Evaluate commercially available reagents for efficiency and cost-effectiveness in suspension culture.
- Step 3: Process Enhancement: Add a lentiviral enhancer reagent to the culture medium post-transfection to boost viral titer. Monitor cell growth and metabolism closely.
- Step 4: Harvest and Clarification: Harvest the viral supernatant approximately 48-72 hours post-transfection. Clarify using depth filtration or centrifugation to remove cell debris.
- Step 5: Downstream Processing and Analytics: Concentrate and purify the clarified supernatant using ultrafiltration/tangential flow filtration (TFF) or chromatography. Key analytics include determining viral titer (e.g., by qPCR for vector genomes) and assessing infectivity (e.g., by flow cytometry on target cells).
Protocol: Formulating and Testing Lipid Nanoparticles (LNPs) for Gene Delivery
This methodology outlines the formulation and in vitro characterization of LNPs, crucial for non-viral gene therapy development [15].
- Step 1: Lipid Mixture Preparation: Prepare an ethanolic solution containing ionizable lipid, phospholipid, cholesterol, and PEG-lipid at a defined molar ratio. The ionizable lipid is critical for endosomal escape.
- Step 2: Aqueous Phase Preparation: Prepare an aqueous buffer containing the nucleic acid payload (e.g., mRNA, siRNA, or plasmid DNA).
- Step 3: Nanoparticle Formation: Rapidly mix the ethanolic lipid solution with the aqueous nucleic acid solution using a microfluidic device or T-mixer. This induces spontaneous formation of LNPs through nanoprecipitation.
- Step 4: Buffer Exchange and Dialysis: Dialyze the formed LNP suspension against a suitable buffer (e.g., PBS) to remove ethanol and establish the final buffer conditions.
- Step 5: Characterization of LNPs:
- Size and Polydispersity: Measure by dynamic light scattering (DLS). Target size is typically 50-200 nm.
- Zeta Potential: Measure surface charge to predict stability and interaction with cell membranes.
- Encapsulation Efficiency: Quantify using a dye exclusion assay (e.g., with Ribogreen for RNA) to determine the percentage of nucleic acid successfully encapsulated.
- In Vitro Transfection Efficiency: Incubate LNPs with target cells and measure gene expression (e.g., via luciferase activity) or gene silencing (via qRT-PCR).
Visualizing Workflows and Challenges
The following diagram illustrates the core workflow and critical challenge points for viral vector manufacturing, highlighting the parallel paths for AAV and Lentivirus production.
The next diagram maps out the development pathway for non-viral vectors, focusing on the design choices and biological barriers that define manufacturing strategy.
The Scientist's Toolkit: Key Research Reagents and Solutions
Successful scale-up requires a suite of reliable tools and reagents. The following table details essential materials used in the development and manufacturing of gene therapy vectors.
Table 3: Essential Research Reagents and Solutions for Vector Manufacturing
| Reagent/Solution | Function in Manufacturing | Example Use Cases |
|---|---|---|
| Suspension Cell Lines | Engineered mammalian cells (e.g., HEK 293) grown in suspension culture to enable scalable production in bioreactors [70]. | Lentiviral and AAV vector production [70]. |
| Chemically Defined Media | Serum-free media that supports high cell density and productivity, enhancing consistency and reducing risk of contamination [70]. | Upstream process for both viral and non-viral production systems [70]. |
| Transfection Reagents | Chemicals or polymers that facilitate the delivery of plasmid DNA into production cells for viral vector generation [70]. | Transient transfection in LV and AV production [70]. |
| Ionizable Lipids | A critical component of LNPs that becomes protonated in acidic endosomes, enabling the release of genetic material into the cytoplasm [9] [15]. | Forming LNPs for mRNA, siRNA, and CRISPR-Cas9 delivery [9] [15]. |
| Polyethyleneimine (PEI) | A cationic polymer used for transfection or as a non-viral gene delivery vector itself, though it can have associated cytotoxicity [15]. | Laboratory-scale transfection and as a benchmark for polymer-based gene delivery [15] [70]. |
| Enhancer Reagents | Additives that boost viral vector titers when added to the production culture, improving overall yield [70]. | Used in lentiviral production platforms to increase functional titer [70]. |
| N-Acetylgalactosamine (GalNAc) | A targeting ligand conjugated to therapeutics to enable receptor-mediated uptake specifically by hepatocytes [14] [3]. | Liver-targeted delivery of siRNA and other RNAi therapeutics [3]. |
The journey to overcome manufacturing complexities in gene therapy is ongoing for both viral and non-viral systems. Viral vectors are tackling challenges of scalability and cost through innovative platforms moving towards high-titer suspension cultures, while non-viral vectors are rapidly evolving to improve efficiency and tissue-specific targeting beyond the liver. The choice between platforms remains a strategic decision, balancing factors of payload size, desired duration of expression, immunogenicity, and ultimately, the feasibility of commercial-scale manufacturing. The continued development of robust, well-characterized, and scalable manufacturing processes is paramount to fulfilling the promise of gene therapies for a broader range of human diseases.
Enhancing Transfection Efficiency and Specificity in Non-Viral Delivery
The field of gene therapy and genetic engineering is increasingly relying on advanced transfection technologies to introduce nucleic acids into cells. While viral vectors have been a traditional mainstay, accounting for 29 of the 35 approved vector-based therapies globally, non-viral delivery systems are gaining significant momentum due to their enhanced safety profiles and manufacturing advantages [3]. The current market analysis indicates substantial growth in this sector, with the non-viral transfection reagents market estimated at USD 745.4 million in 2025 and projected to reach USD 1319.5 million by 2032, reflecting a compound annual growth rate of 8.5% [71]. This growth is largely driven by increasing demand for gene and cell therapies to treat genetic disorders, cancer, and other life-threatening conditions, pushing the need for efficient non-viral transfection systems that can overcome the limitations of viral approaches, including immunogenicity, insertional mutagenesis, and limited packaging capacity [25] [71].
Non-viral vectors encompass a broad category of synthetic delivery systems, including lipid-based nanoparticles, polymeric nanoparticles, peptide-based carriers, and inorganic materials, as well as physical delivery methods [72]. These systems must overcome multiple biological barriers to achieve successful transfection, including protection from nucleases, passage through the negatively charged cell membrane, escape from endosomal compartments, navigation through the cytoplasmic matrix, and for DNA delivery, translocation across the nuclear membrane [72]. The continuous evolution of these technologies is pivotal for advancing gene and cell-based therapies, with innovations in non-viral delivery platforms offering safer and more adaptable alternatives to viral vectors [73]. This comparison guide provides an objective analysis of non-viral transfection performance relative to viral methods, with supporting experimental data and detailed methodologies to inform research and development decisions.
Comparative Analysis: Viral vs. Non-Viral Delivery Systems
Fundamental Characteristics and Performance Metrics
The choice between viral and non-viral delivery systems involves careful consideration of multiple factors, including transfection efficiency, safety profile, cargo capacity, and manufacturing feasibility. The table below provides a comprehensive comparison of these fundamental characteristics:
| Characteristic | Viral Vectors | Non-Viral Vectors |
|---|---|---|
| Origin | Derived from viruses (e.g., AAV, Lentivirus, Adenovirus) | Synthetic (e.g., lipids, polymers, peptides) [72] |
| Transfection Efficiency | High (evolved natural infection mechanisms) | Variable; typically lower but improving with advanced LNPs and optimization [25] [72] |
| Immune Response | Significant concern (can trigger innate and adaptive immunity) | Greatly reduced immunogenicity [25] [72] |
| Integration Risk | Yes (random insertion with retroviruses/LVs) | Minimal to none (non-integrating) [25] [72] |
| Cargo Capacity | Limited (AAV: ~4.7 kb; Lentivirus: ~8 kb) [3] | Large (up to 22 kb; can deliver mRNA) [72] |
| Manufacturing Scalability | Complex, expensive production | Generally simpler, more scalable, cost-effective [25] |
| Toxicity/Cytotoxicity | Variable; depends on viral system | Low cytotoxicity with optimized formulations [71] |
| Clinical Applications | In vivo therapy (e.g., Luxturna, Zolgensma), ex vivo cell engineering (CAR-T) [3] | In vivo RNA/siRNA therapy (e.g., Onpattro, mRNA vaccines), ex vivo cell engineering [3] |
Quantitative Performance Comparison of Non-Viral Methods
Different non-viral transfection methods exhibit varying efficiencies across cell types. A study optimizing gene delivery to the Mehr-80 human large cell lung cancer cell line provides comparative efficiency data and cytotoxicity assessment:
| Transfection Method | Mechanism | Transfection Efficiency | Key Optimization Parameters | Cytotoxicity Notes |
|---|---|---|---|---|
| Lipofection (Lipofectamine 2000) | Chemical; cationic lipid-DNA complexes | 40.1% (GFP+ cells at 48 hr) [74] | DNA (µg) to reagent (µl) ratio; 90% cell confluency [74] | Low cytotoxicity observed [71] |
| Electroporation | Physical; electrical pulses create membrane pores | 34.1% (GFP+ cells at 48 hr) [74] | Field strength, pulse duration, buffer ionic strength [74] | Cell viability impact requires optimization [74] |
| Calcium Phosphate (CaP) | Chemical; precipitate formation and endocytosis | 8.4% (GFP+ cells at 48 hr) [74] | Precipitate formation time; glycerol shock application [74] | N/A |
| DEAE-Dextran | Chemical; cationic polymer-DNA complexes | 8.2% (GFP+ cells at 48 hr) [74] | Polymer concentration; DMSO shock application [74] | N/A |
| Superfect | Chemical; activated dendrimer | 4.9% (GFP+ cells at 48 hr) [74] | Incubation time; DNA to reagent ratio [74] | N/A |
| Polyethylenimine (PEI) | Chemical; cationic polymer ("proton sponge" effect) | High in optimized conditions (e.g., T cells) [75] | N/P ratio; cell density; extracellular media pH/osmolarity [76] [75] | Cytotoxicity concerns, especially with high molecular weight PEI [25] |
Optimization Parameters for Enhanced Non-Viral Transfection
Vector Design and Formulation Parameters
The efficiency of non-viral transfection is heavily influenced by the physicochemical properties of the delivery vector. Key design parameters that require optimization include:
Composition and Charge: Cationic lipids or polymers are typically used to encapsulate nucleic acids through electrostatic interactions, but highly positive charges can increase toxicity. A balance must be struck between efficient complexation and biocompatibility. Ionizable lipids, which are positively charged at low pH (aiding encapsulation) and neutral at physiological pH (reducing toxicity), have been crucial for clinical success, as seen in LNPs used for mRNA vaccines and Onpattro [72]. Surface modification with polyethylene glycol (PEG) creates a "stealth" effect, diminishing interaction with serum proteins and enhancing stability and circulation time [72].
Size and Morphology: Nanoparticle size significantly impacts cellular uptake pathways and overall efficiency. Optimal sizes generally range from 60-100 nm, as particles smaller than 50 nm are quickly cleared by the kidneys, while those larger than 300 nm tend to activate immune responses [72]. The shape of nanoparticles also influences internalization, with rod-shaped geometries potentially offering superior cellular uptake compared to spherical particles [72].
Surface Functionalization: The addition of targeting ligands (e.g., antibodies, peptides, carbohydrates, or aptamers) to the vector surface enables cell-specific delivery, improving specificity and reducing off-target effects [77]. This strategy is exemplified by GalNAc conjugation for liver-targeted delivery of RNA therapies, which has enabled multiple FDA-approved drugs including givosiran, lumasiran, and inclisiran [3].
Experimental and Cellular Optimization Strategies
Beyond vector design, experimental conditions and cellular parameters significantly influence transfection outcomes. Research with human mesenchymal stem cells (hMSCs) and T cells has identified several critical factors:
Cell Density and Proliferation State: Studies on hMSCs demonstrated that culture conditions inhibiting cell division also decreased transfection efficiency, suggesting that strategies promoting cell proliferation may enhance transfection [76]. Optimal cell density at the time of transfection is also critical, with both too sparse and too confluent cultures potentially reducing efficiency [76].
N/P Ratio and Polyplex Dose: The N/P ratio, defined as the ratio of primary amines on the cationic polymer (e.g., PEI) to phosphate groups on the nucleic acid, significantly influences transfection efficiency and cytotoxicity [76]. For PEI-mediated transfection of T cells, an N/P ratio of 8.0 was found to be optimal, with further improvements achievable through adjustments in DNA dosage and complex volume [75].
Novel Transfection Protocols: Innovative approaches to the transfection process itself can yield substantial improvements. For T cell transfection, reversing the protocol by performing transfection in vials rather than in culture plates resulted in a 20-fold increase in cellular uptake and transfection efficiency compared to conventional direct transfection [75]. Similarly, modifying cellular physiology with hypotonic extracellular media at pH 9.0 dramatically enhanced transfection rates while maintaining minimal cytotoxicity [75].
Advanced Strategies for Targeted Delivery
Cell-Specific Targeting Approaches
Achieving cell-type specificity remains a significant challenge in non-viral gene delivery. Two primary strategies have emerged to address this limitation:
Ligand-Mediated Targeting: Delivery vectors can be modified with cell-specific ligands that recognize and bind to receptors on target cells. These include natural ligands, antibodies, peptides, carbohydrates, or aptamers that enable targeted uptake with higher specificity and improved biodistribution [77]. For example, transferrin-polyethylene glycol-poly-l-lysine (Tf-PEG-PLL) conjugates can target cells expressing transferrin receptors, while folate-conjugated systems target folate receptor-overexpressing cells [77].
Route-Specific Administration: The administration pathway significantly influences biodistribution and cellular uptake. Different routes—including mucosal, intravenous, intramuscular, subcutaneous, intradermal, intratumoral, and intraocular—can be selected to best facilitate optimal uptake into targeted cells and organs [77]. Intravenous administration often leads to predominant liver accumulation, while localized administration (e.g., intratumoral, intraocular) can enhance delivery to specific tissues.
Experimental Protocols for Enhanced Transfection
Protocol: Optimization of PEI-Mediated Transfection in Human T Cells
This protocol, adapted from research demonstrating a 20-fold improvement in transfection efficiency, outlines key optimization steps for challenging-to-transfect immune cells [75]:
- Nanoparticle Preparation: Form PEI/DNA nanoparticles at an N/P ratio of 8.0 in 150 mM sterile NaCl. Allow complexation for 25 minutes at room temperature.
- Cell Preparation: Culture T cells in appropriate medium. Increase cell seeding density significantly above conventional recommendations.
- Reverse Transfection in Vials: Perform transfection in sealed vials rather than culture plates to enhance nanoparticle-cell interaction.
- Media Adjustment: Shortly after transfection, add complete media to support cell viability and recovery.
- Cellular Physiology Modulation: Treat cells with hypotonic extracellular media adjusted to pH 9.0 during transfection to enhance membrane permeability and nanoparticle uptake.
- Post-Transfection Processing: After 4-6 hours, transfer cells to standard culture conditions and assess transfection efficiency at 24-48 hours.
This optimized protocol demonstrates how methodological innovations can substantially improve non-viral gene delivery to even recalcitrant primary cell types [75].
Protocol: Lipid Nanoparticle Formulation for mRNA Delivery
The success of LNPs in clinical applications relies on precise formulation parameters:
- Lipid Composition: Combine ionizable lipids, phospholipids, cholesterol, and PEG-lipids in specific molar ratios (typically 50:10:38.5:1.5). The ionizable lipid is critical for encapsulation and endosomal escape.
- mRNA Preparation: Dilute mRNA in aqueous buffer at appropriate concentration.
- Rapid Mixing: Use microfluidic devices to rapidly mix aqueous mRNA solution with lipid solution in ethanol, enabling spontaneous nanoparticle formation.
- Buffer Exchange and Concentration: Dialyze or diafilter against physiological buffer to remove ethanol and concentrate the final LNP product.
- Characterization: Assess particle size (optimally 60-100 nm), polydispersity, encapsulation efficiency, and stability before use.
This methodology has proven crucial for clinical applications, including mRNA vaccines and siRNA therapeutics like Patisiran [72] [3].
Essential Research Reagent Solutions
Successful transfection experiments require specific reagents and systems optimized for different applications. The following table details key research solutions and their functions:
| Reagent/System | Function | Application Notes |
|---|---|---|
| Lipofectamine 2000 | Cationic lipid reagent forms complexes with nucleic acids for cellular uptake | High efficiency for many cell lines; optimized for plasmid DNA and siRNA [74] |
| Polyethylenimine (PEI) | Cationic polymer condenses DNA/RNA via electrostatic interactions; "proton sponge" effect promotes endosomal escape | Cost-effective for large-scale transfections; branching and molecular weight affect efficiency/toxicity [76] [75] |
| Ionizable Lipids | pH-dependent charge enables RNA encapsulation and endosomal escape while reducing toxicity | Core component of clinical LNPs (e.g., Onpattro, COVID-19 vaccines) [72] |
| Superfect | Activated dendrimer forms stable complexes with DNA | Suitable for difficult-to-transfect cells; requires optimization of DNA:reagent ratio [74] |
| Electroporation Systems | Electrical pulses create temporary pores in cell membranes for nucleic acid entry | Effective for hard-to-transfect cells (e.g., primary cells, immune cells); parameters require optimization [74] |
| GalNAc Conjugates | Targets asialoglycoprotein receptor on hepatocytes for liver-specific delivery | Enables subcutaneous administration of RNA therapies; used in multiple approved drugs [3] |
| Cell-Specific Ligands | Antibodies, peptides, or aptamers conjugated to vectors for targeted delivery | Enhances specificity; reduces off-target effects [77] |
The field of non-viral transfection continues to evolve rapidly, with current research addressing remaining challenges in efficiency, specificity, and applicability beyond hepatic tissues. Innovations in vector design, including novel ionizable lipids, biodegradable polymers, and hybrid systems, are steadily closing the efficiency gap with viral vectors while maintaining superior safety profiles [73] [77]. The integration of advanced targeting strategies and refined administration approaches is expanding the potential applications of non-viral systems to previously challenging targets, including extrahepatic tissues and specific cell populations within complex organs [77] [3].
The growing clinical validation of non-viral platforms, particularly lipid nanoparticles for mRNA and siRNA delivery, has established a robust foundation for future therapeutic development. As the understanding of intracellular trafficking mechanisms deepens and material science advances, next-generation non-viral vectors are poised to offer unprecedented precision in gene delivery [73] [77]. These innovations, combined with manufacturing advantages and favorable safety profiles, position non-viral transfection technologies as increasingly central to the future of gene therapy, regenerative medicine, and personalized treatments [73] [71]. For research and development applications, the systematic optimization of vector parameters, cellular conditions, and transfection protocols outlined in this guide provides a pathway to maximize transfection efficiency and specificity for diverse experimental and therapeutic objectives.
The development of somatic cell gene therapies is propelled by parallel innovations in viral and non-viral delivery platforms. Viral vectors, particularly Adeno-Associated Viruses (AAV), are being refined through advanced capsid engineering to enhance targeting and safety. In parallel, non-viral delivery, led by Lipid Nanoparticles (LNPs), is being transformed by novel ionizable lipids that improve efficacy and reduce toxicity. Underpinning both fields is a critical push toward automation and modernized manufacturing to overcome scalability and cost barriers. The following guide provides a detailed, data-driven comparison of these technologies to inform therapeutic development strategies.
Viral Vector Innovations: Engineering the Capsid
Core Strategic Approaches
The natural tropism of AAV serotypes often lacks the specificity and efficiency required for advanced therapies, driving the development of engineered capsids. Three primary methodologies are employed [78]:
- Rational Design: Leverages known capsid structure-activity relationships to make targeted modifications. This approach is systematic but requires deep mechanistic insight.
- Directed Evolution: Uses iterative rounds of selection on high-diversity random peptide libraries to identify variants with desired properties in an unbiased manner.
- Machine Learning (ML)-Guided Design: Analyzes high-throughput screening data to generate predictive models that accelerate the discovery of novel capsids, integrating the benefits of both rational design and directed evolution.
Experimental Workflow for Capsid Engineering
A modern, integrated workflow for developing novel AAV capsids is exemplified by the Barcoded Rational AAV Vector Evolution (BRAVE) platform [79]. The methodology below can be adapted for various target cell types.
Key Experimental Steps [79]:
- Library Construction: A library of AAV capsid variants is generated by inserting degenerate oligonucleotides into surface-exposed loops of the VP1 capsid protein, creating millions of potential variants.
- Barcoding: A unique DNA barcode is inserted into the non-coding region of the packaged genome for each capsid variant, enabling high-throughput tracking.
- Parallel Screening: The pooled library is administered to the target system (e.g., human glial spheroids, animal models). The BRAVE platform emphasizes parallel screening across different species and tissues to assess translatability early.
- Recovery & Sequencing: Viral DNA is recovered from target tissues, and the barcodes are amplified and sequenced via NGS to quantify the enrichment of specific capsids.
- Validation: Lead candidates are produced as individual vectors and rigorously validated for transduction efficiency, specificity, and packaging capability in relevant in vitro and in vivo models.
Non-Viral Vector Innovations: Designing the Lipid
The Anatomy of a Modern LNP
LNPs used for nucleic acid delivery are complex, multi-component systems. The core structure and function of each component are summarized below [80] [72]:
- Ionizable Lipid: The most critical functional component. It is positively charged at low pH to enable RNA complexation and endosomal escape, but neutral at physiological pH to reduce toxicity.
- Phospholipid (e.g., DSPC): Contributes to the structural integrity of the LNP bilayer and promotes fusion with the endosomal membrane.
- Cholesterol: Stabilizes the LNP structure and enhances cellular uptake by increasing membrane fluidity.
- PEGylated Lipid: Shields the LNP surface to reduce nonspecific interactions with serum proteins, prolongs circulation time, and prevents particle aggregation.
Development Workflow for Novel Lipids
The discovery of novel ionizable lipids is a structured process that bridges computational chemistry, in vitro screening, and in vivo validation.
Key Experimental Steps [80]:
- Lipid Synthesis & Library Design: A library of ionizable lipids is synthesized based on rational design principles, often exploring diverse headgroups, linkers, and hydrocarbon tails.
- High-Throughput In Vitro Screening: LNPs are formulated with each novel lipid and a reporter mRNA (e.g., luciferase). Key performance metrics are measured:
- Encapsulation Efficiency: Typically quantified using a Ribogreen assay, with >90% being a target for clinical-grade LNPs.
- Expression/Potency: Measured by luciferase activity (RLU/mg protein) in relevant cell lines (e.g., HepG2 for liver tropism).
- Cytotoxicity: Assessed via assays like CellTiter-Glo to ensure cell viability >80% at therapeutic doses.
- Lead Optimization & In Vivo Validation: Top-performing lipids are selected for further formulation optimization. The final LNP candidates are then evaluated in animal models to confirm biodistribution, therapeutic efficacy, and tolerability.
Comparative Performance Data
Technology Feature Comparison
The choice between viral and non-viral systems involves fundamental trade-offs, as detailed in the table below [38] [26] [72].
| Feature | AAV (Viral) | Lentivirus (Viral) | LNP (Non-Viral) |
|---|---|---|---|
| Primary Use Case | In vivo gene replacement (CNS, eye, liver) [38] | Ex vivo cell therapy (CAR-T, HSCs) [38] | Gene editing (CRISPR), vaccines, transient expression [38] |
| Cargo Capacity | ~4.7 kb (strict) [38] | ~10 kb (moderate) [38] | Flexible / High (suitable for mRNA) [72] |
| Genetic Persistence | Episomal (long-term in non-dividing cells) [38] | Integrated (permanent in dividing cells) [38] | Transient (ideal for editing) [38] |
| Immunogenicity | High (pre-existing antibodies, no re-dosing) [38] | Low (mostly used ex vivo) [38] | Low (re-dosable) [38] |
| Key Innovation | Capsid Engineering (tropism, evasion) [78] | Self-Inactivating (SIN) Vectors (safety) [24] | Novel Ionizable Lipids (efficiency, toxicity) [80] |
| Manufacturing COGS | High (complex culture & purification) [38] | High (shear sensitivity, low yield) [38] | Low to Medium (chemical synthesis) [38] |
Quantitative Manufacturing Metrics
Modernization efforts are significantly impacting the production economics of both platforms [81] [82].
| Manufacturing Metric | Legacy Viral Vector Process | Advanced Automated Process | LNP Manufacturing |
|---|---|---|---|
| Upstream Method | Transient Transfection (HEK293) | Stable Producer Cell Lines [81] | Microfluidic Mixing [38] |
| Primary Cost Driver | Plasmid DNA & Purification [81] | Cell Line Development [81] | Lipid Raw Material Purity [38] |
| Key Bottleneck | Empty/Full Capsid Separation [38] | Viral Stability & Titer [38] | Lipid Purity & Microfluidic Fouling [38] |
| Batch Duration | Weeks [81] | Weeks (but less hands-on) | Days [38] |
| Scalability | Challenging, moving to suspension bioreactors [81] | Improved with fixed-bed bioreactors (LV) & suspension (AAV) [81] | Highly Scalable [38] |
The Scientist's Toolkit: Essential Research Reagents
Successful development in this field relies on a suite of specialized reagents and tools. The following table details key solutions for core experimental workflows.
| Research Reagent / Solution | Primary Function | Example Application |
|---|---|---|
| Ionizable Lipids (e.g., DLin-MC3-DMA, SM-102) | Enable mRNA encapsulation and endosomal escape in LNPs [80]. | Formulating LNP-based mRNA vaccines or therapeutics. |
| Stable Producer Cell Lines | Stably express viral components, eliminating need for plasmid transfection [81]. | Scalable, high-yield AAV or Lentivirus production. |
| Synthetic DNA | Enzymatically produced starting material; avoids bacterial fermentation impurities [81]. | More consistent and scalable viral vector production. |
| Molecular Barcodes | Unique DNA sequences for tracking individual capsid variants in a pool [79]. | High-throughput screening in AAV capsid engineering. |
| PEGylated Lipids | Shield LNPs, reduce protein adsorption, and increase circulation half-life [80] [72]. | Improving the pharmacokinetics and stability of LNP formulations. |
| Capsid Library Plasmids | Engineered plasmids with regions for peptide insertion to generate capsid diversity [78] [79]. | Creating diverse AAV libraries for directed evolution. |
The data and methodologies presented underscore that the choice between viral and non-viral delivery is not a matter of superiority, but of strategic alignment with therapeutic goals.
- Choose AAV when the goal is durable expression in static tissues (e.g., CNS, retina, liver) and the genetic cargo is within its ~4.7 kb limit. Invest in capsid engineering to overcome tropism and immunogenicity barriers [38] [78].
- Choose Lentivirus for ex vivo cell engineering (e.g., CAR-T, HSCs) where permanent integration in dividing cells is required. Leverage SIN designs to mitigate insertional mutagenesis risks [38] [24].
- Choose LNPs for transient expression applications, such as delivering CRISPR-based gene editors or vaccines, or when re-dosing is anticipated. Focus on novel ionizable lipid designs to improve tissue targeting beyond the liver and enhance endosomal escape efficiency [38] [80].
The future of somatic cell gene therapy lies in a "delivery-agnostic" pipeline, where the optimal vector is selected based on the specific indication, target tissue, and payload requirements. Continued innovation in capsid engineering, lipid chemistry, and—critically—manufacturing technology will be the key drivers in making these transformative therapies more effective, safe, and accessible.
Direct Comparative Analysis: Evaluating Vector Performance and Selection Criteria
The choice of a delivery vector is a foundational decision in genetic medicine, critically influencing the therapeutic outcome, safety, and ultimate clinical success of a product. This guide provides an objective, data-driven comparison between viral and non-viral vector platforms, focusing on their distinct safety profiles and immunogenicity. These characteristics are paramount for researchers and drug development professionals designing new therapies, particularly in the context of Somatic Cell Gene Therapy (SMGT). Viral vectors, born from engineered viruses, leverage millions of years of evolutionary optimization for gene delivery, often resulting in high transduction efficiency and durable expression [25]. In contrast, non-viral vectors, which include synthetic molecules like lipids and polymers, offer a more controlled and scalable manufacturing process, typically with a superior initial safety profile [38] [25]. The landscape is rapidly evolving; the mantra was once "AAV for in vivo, Lentivirus for ex vivo," but the rise of CRISPR-based editing and proven scalability of platforms like Lipid Nanoparticles (LNPs) is compelling a thorough re-evaluation of this dogma [38]. This guide synthesizes current evidence to inform strategic platform selection, framing the discussion within the critical trade-offs between efficacy, safety, and manufacturability.
Comparative Analysis of Vector Platforms
The following tables provide a consolidated overview of the core characteristics, safety, and immunogenicity profiles of the leading vector platforms.
Table 1: Platform Characteristics and Clinical Applications
| Feature | AAV (Viral) | Lentivirus (Viral) | LNP (Non-Viral) |
|---|---|---|---|
| Primary Use Case | In vivo gene replacement (CNS, Eye, Liver) [38] | Ex vivo cell therapy (CAR-T, HSCs) [38] [3] | Gene editing (CRISPR/mRNA), Vaccines [38] |
| Cargo Capacity | ~4.7 kb (Strict) [38] [83] | ~10 kb (Moderate) [38] | Flexible / High [38] |
| Genetic Persistence | Episomal (Long-term in non-dividing cells) [38] [83] | Integrated (Permanent in dividing cells) [38] [3] | Transient (Ideal for editing) [38] |
| Key Manufacturing Bottleneck | Empty/Full Capsid Separation [38] | Viral Stability & Titer [38] | Lipid Purity & Microfluidic Fouling [38] |
Table 2: Safety and Immunogenicity Profile Comparison
| Profile | AAV (Viral) | Lentivirus (Viral) | LNP (Non-Viral) |
|---|---|---|---|
| Immunogenicity | High (Pre-existing NAbs prevent re-dosing) [38] [84] | Low (Use is mostly ex vivo) [38] | Low (Re-dosable, though PEG-antibodies are a concern) [38] |
| Primary Safety Concerns | Pre-existing immunity; inflammatory responses; rare serious adverse events (e.g., VITT with some adenovirus vectors) [84] | Insertional mutagenesis (theoretical risk of oncogenesis) [38] [3] | Reactogenicity (e.g., injection site pain, fatigue, headache); biodistribution largely to the liver [85] [38] |
| Typical Local Adverse Events | Injection-site pain (20-64% in rVSV-ΔG-spike vaccine trial) [85] [86] | Varies by application (ex vivo) | Injection site tenderness/pain (e.g., 27-71% in mRNA RSV vaccine trial) [87] |
| Typical Systemic Adverse Events | Fatigue (21-33%), Headache (15-22%) [85] [86] | Varies by application (ex vivo) | Irritability, loss of appetite, sleepiness (most Grade 1/2) [87] |
| Re-dosability | Not feasible due to anti-vector immunity [38] | Feasible for ex vivo applications | Feasible, does not generate strong anti-vector immunity [38] |
Experimental Protocols for Profiling Vectors
To generate the comparative data presented above, standardized experimental protocols are employed. The following workflows detail key methodologies for assessing immunogenicity and safety in preclinical and clinical studies.
Preclinical Immunogenicity and Safety Workflow
Title: Preclinical safety and immunogenicity workflow.
Detailed Methodology:
- In Vitro Models: Utilize human cell lines or primary peripheral blood mononuclear cells (PBMCs) to conduct preliminary assessments of vector-induced innate immune activation (e.g., cytokine release) and cytotoxicity [25].
- Humoral Response Profiling: A critical step for viral vectors. This involves screening for Pre-existing Neutralizing Antibodies (NAbs) using serum from animal models or human donors. Assays measure the ability of serum antibodies to block vector transduction of permissive cells in vitro [38] [84]. Post-administration, Anti-transgene Antibodies are quantified via ELISA to assess unwanted humoral immunity against the therapeutic protein [83].
- Cellular Immune Response Assays: Flow cytometry is used to characterize T-cell responses, particularly the activation of CD8+ Cytotoxic T Lymphocytes (CTLs) and CD4+ T-helper cells against the transgene product or viral capsid. An unwanted, robust CTL response can lead to the elimination of transfected cells [84].
- In Vivo Animal Studies: Selected animal models are dosed with the vector candidate. The choice of model is critical, as immunogenicity in animal models does not always accurately predict human responses [25].
- Biodistribution Study: Tissues are collected post-mortem. Quantitative polymerase chain reaction (qPCR) is performed on DNA extracts from organs like the liver, spleen, and heart to quantify vector genome copies and determine the vector's distribution and potential off-target accumulation [3].
- Toxicology & Histopathology: A comprehensive analysis where tissues are examined grossly and microscopically for signs of toxicity, inflammation, or other pathology related to the vector administration [85].
Clinical Phase Immunogenicity Assessment
Title: Clinical immunogenicity assessment workflow.
Detailed Methodology:
- Participant Enrollment & Pre-dose Serotyping: Before dosing, participants are screened for pre-existing immunity to the vector (e.g., anti-AAV or anti-adenovirus NAbs). This is a key enrollment criterion, as high titers often lead to exclusion [38] [84].
- Serial Blood Collection: Blood is drawn at baseline and multiple post-administration timepoints (e.g., Day 14, 28, 56) to track the evolution of the immune response [85] [86].
- Neutralizing Antibody (NAb) Assay: This functional assay measures serum's capacity to neutralize a live, replicating vector or a pseudovirus, providing a direct measure of potentially immunity-blocking antibodies [85] [87].
- Binding Antibody Assay: Techniques like Enzyme-Linked Immunosorbent Assay (ELISA) or Meso Scale Discovery (MSD) are used to quantify total antibody titers (e.g., IgG) against the transgene product (e.g., spike protein) or specific viral capsid proteins [86].
- Cellular Immunophenotyping: Intracellular cytokine staining (ICS) and Enzyme-Linked ImmunoSpot (ELISpot) assays are performed on PBMCs to quantify antigen-specific T-cells producing cytokines like IFN-γ [84].
- Data Analysis: Immunogenicity is expressed quantitatively. Geometric Mean Titer (GMT) summarizes antibody levels across groups. Geometric Mean Fold Rise (GMFR) compares post-vaccination to pre-vaccination titers. Seroconversion is defined as a pre-specified fold-increase in antibody titer (e.g., ≥4-fold) from baseline [85] [87].
Essential Research Reagent Solutions
The following table catalogues key reagents and their applications in vector research and characterization, serving as a toolkit for experimental design.
Table 3: Key Research Reagents for Vector Analysis
| Reagent / Assay | Primary Function | Application Context |
|---|---|---|
| Cationic Lipids (e.g., CD-Chol) | Form lipoplexes with nucleic acids via electrostatic interactions; facilitate cellular uptake and endosomal escape [25]. | Formulating non-viral vectors (LNPs) for in vivo delivery of mRNA or CRISPR components [38] [25]. |
| Polyethylenimine (PEI) | A cationic polymer that condenses genetic material, induces endosomal burst via the "proton sponge" effect [25]. | A gold-standard transfection reagent in vitro; explored for in vivo non-viral gene delivery despite cytotoxicity concerns [25]. |
| VSV-G Pseudotyped Lentivirus | The Vesicular Stomatitis Virus Glycoprotein (VSV-G) confers broad tropism and enhances viral particle stability [84]. | Standard for producing high-titer lentiviral vectors for ex vivo cell transduction (e.g., CAR-T therapy) [84] [3]. |
| Anti-AAV Neutralizing Antibody Assay | Measures levels of pre-existing antibodies that block AAV transduction, using live virus or reporter systems [38] [84]. | Critical for patient screening in AAV-based clinical trials to exclude those with high NAb titers [38] [84]. |
| ELISpot / Intracellular Cytokine Staining | Detects and quantifies antigen-specific T-cells (e.g., against transgene or capsid) at the single-cell level [84]. | Assessing cell-mediated immunogenicity in preclinical models and clinical trial samples [84]. |
| Anion-Exchange Chromatography Resins | Separates viral particles based on surface charge differences; crucial for resolving full capsids from empty capsids [38]. | Downstream purification of AAV vectors; a major bottleneck in manufacturing. New resins improve separation resolution [38]. |
The choice between viral and non-viral vectors is not a binary one but a strategic decision based on the therapeutic target. Viral vectors, particularly AAV and Lentivirus, remain powerful for applications requiring high transduction efficiency and long-lasting gene expression, but they are hampered by immunogenicity, pre-existing immunity, and complex manufacturing [38] [84] [25]. Non-viral vectors, especially LNPs, offer a compelling alternative with their safety, re-dosability, and rapid, scalable production, though they face challenges in biodistribution and delivery efficiency beyond the liver [38] [3]. The future lies in platform-agnostic pipelines and hybrid approaches. Innovations like engineered capsids to evade NAbs, novel ionizable lipids for extra-hepatic targeting, and the treatment of delivery systems as regulatable platforms are accelerating the field [38] [84]. For researchers, the guiding principle remains selecting the vector that delivers the optimal therapeutic index for the patient, ensuring that groundbreaking genetic therapies can be safely, effectively, and scalably manufactured.
Gene therapy's success is fundamentally governed by the efficiency of its delivery vectors, which are broadly categorized into viral and non-viral systems. For researchers and drug development professionals, selecting the optimal vector requires a careful balance between transduction efficiency—the proportion of cells successfully expressing the transgene—and the duration of transgene expression. Viral vectors, honed by evolution, typically offer high transduction efficiency and sustained expression but can pose safety risks such as immunogenicity and insertional mutagenesis [31] [25]. Non-viral vectors, conversely, present a more favorable safety profile and greater design flexibility but often struggle with lower transfection efficiency and transient expression patterns [3] [24]. This guide provides a data-driven comparison of these platforms, summarizing key efficiency metrics in structured tables and detailing the experimental protocols used to generate them, to inform strategic decisions in therapeutic development.
Comparative Performance Analysis of Vector Systems
The core metrics for evaluating gene delivery vectors are summarized in the table below, which synthesizes data from current literature and clinical applications.
Table 1: Comparative Efficiency Metrics of Viral and Non-Viral Vectors
| Vector Type | Typical Transduction Efficiency | Duration of Expression | Key Advantages | Primary Limitations |
|---|---|---|---|---|
| Lentivirus (LV) | 30-70% (ex vivo T-cells) [31] | Long-term (stable genomic integration) [24] | Transduces dividing & non-dividing cells; stable expression [25] | Risk of insertional mutagenesis [3] |
| Adeno-associated Virus (AAV) | Varies by serotype/route (e.g., robust CNS with ICV delivery) [88] | Long-term (persistent episomes) [3] | Low immunogenicity; good safety profile [3] [89] | Limited cargo capacity (~4.7 kb); pre-existing immunity [3] |
| Adenovirus (AdV) | High [31] | Short-term (transient, non-integrating) [31] | Large cargo capacity; high titer production [24] | High immunogenicity; strong inflammatory response [3] [89] |
| Lipid Nanoparticles (LNP) | Lower than viral vectors [3] | Short-term (transient expression) [89] | Low immunogenicity; redosing possible; scalable manufacturing [89] | Lower transfection efficiency; mostly liver biodistribution [3] |
| GalNAc Conjugates | High for hepatocytes [1] | Transient (requires redosing) [3] | Excellent liver tropism; subcutaneous administration [3] | Primarily restricted to liver applications [3] |
Beyond these core metrics, the Vector Copy Number (VCN), or the average number of vector integrations per cell, is a critical quality attribute for integrating vectors like Lentivirus. Clinical programs generally maintain a VCN below 5 copies per cell to balance therapeutic efficacy against genotoxic risks [31]. Another crucial consideration is biodistribution, which is heavily influenced by the administration route. For example, a 2025 study on AAV9 demonstrated that intracerebroventricular (ICV) delivery produced robust transduction in the central nervous system (CNS), while intravenous (IV) administration led to widespread peripheral organ expression with limited CNS penetration [88].
Key Experimental Protocols for Efficiency Evaluation
Robust experimental design is essential for the accurate evaluation of vector performance. The following protocols are standard in the field for assessing transduction rates and expression durability.
Protocol 1: Evaluating Transduction Efficiency in T-Cells Using Flow Cytometry
This ex vivo protocol is critical for cell therapies, such as CAR-T manufacturing [31] [90].
- T-Cell Activation: Isolate human Peripheral Blood Mononuclear Cells (PBMCs) from donor blood. Activate the T-cells by culturing them for 3 days in a medium supplemented with a CD3/CD28/CD2 T-cell activator and interleukin-2 (IL-2, typically 50 IU/mL) [90].
- Viral Transduction: On day 0, pellet the activated PBMCs and resuspend them in a fresh medium containing the viral vector (e.g., Lentivirus encoding a GFP transgene). The vector is used at a specific Multiplicity of Infection (MOI), which is the ratio of infectious vector particles to target cells. A common method involves static incubation in a 24-well plate [90].
- Post-Transduction Culture: After 24 hours, harvest the cells, wash them to remove excess vector, and reseed them in a fresh growth medium containing IL-2. Culture the cells for an additional 3-5 days to allow for transgene expression [90].
- Efficiency Analysis: On day 4 or 5, analyze the cells by flow cytometry. Cells are typically stained with a viability dye (e.g., Viobility 405/452 Fixable Dye) and an antibody against CD3 to identify T-cells. Transduction efficiency is calculated as the percentage of live, CD3+ cells that are positive for the transgene (e.g., GFP) [31] [90].
Protocol 2: Assessing In Vivo Transduction and Duration in Liver
This in vivo protocol is used for liver-directed gene therapies, as seen in studies for hemophilia [91].
- Vector Administration: Systemically administer the vector (e.g., LV or AAV) into experimental mice via intravenous (IV) injection. The dose is measured in vector genomes per kilogram of body weight (vg/kg). To enhance transduction a priori, mice may be pre-treated with regimens such as fasting or transient inhibition of antiviral pathways (e.g., using an anti-IFNAR1 antibody) [91].
- Long-Term Monitoring & Selection: For integrating vectors, an a posteriori selection strategy can be employed to enhance potency. This involves treating mice with a hepatotoxic drug like acetaminophen (paracetamol) several weeks after transduction. This selectively eliminates non-transduced hepatocytes (with normal metabolic activity), thereby enriching for the genetically corrected population and increasing overall transgene output over time [91].
- Output Measurement: Regularly collect blood samples over weeks or months. Quantify the concentration of the secreted transgene product (e.g., human Factor IX or Factor VIII for hemophilia models) in the plasma using an enzyme-linked immunosorbent assay (ELISA) [91].
- Terminal Analysis: At the end of the study, euthanize the animals and harvest tissues. Analyze the liver for:
- Vector Copy Number (VCN): Using droplet digital PCR (ddPCR) on extracted genomic DNA to quantify the number of integrated vector genomes per cell [31] [91].
- Transgene Expression: Via immunohistochemistry or Western blotting on tissue lysates.
- Biodistribution: Measure VCN in other organs (e.g., spleen, heart, lungs) to assess off-target transduction [88].
The logical workflow for evaluating gene delivery systems, from initial vector preparation to final data analysis, is outlined in the following diagram:
The Scientist's Toolkit: Essential Research Reagents
Successful execution of these protocols relies on a suite of critical reagents and instruments. The following table details key solutions for vector production and evaluation.
Table 2: Key Research Reagent Solutions for Gene Delivery Evaluation
| Item Name | Function/Application | Example Use Case |
|---|---|---|
| Lentiviral Vectors (VSV-G pseudotyped) | Efficient gene delivery to broad cell types, including non-dividing cells [31]. | Ex vivo T-cell engineering (CAR-T therapy) [90]. |
| AAV Vectors (various serotypes) | In vivo gene delivery with serotype-specific tissue tropism (e.g., AAV9 for CNS) [88]. | Gene therapy for neurological disorders via ICV injection [88]. |
| Interleukin-2 (IL-2) | T-cell growth factor; enhances survival and expansion post-transduction [31]. | Added to culture medium for ex vivo T-cell transduction and expansion [90]. |
| CD3/CD28 T-Cell Activator | Mimics antigen presentation, activating T-cells and upregulating receptors for transduction [90]. | Pre-activation of PBMCs before viral transduction to improve efficiency [31]. |
| Droplet Digital PCR (ddPCR) | Absolute quantification of Vector Copy Number (VCN) with high precision [31]. | Measuring the number of viral integrations in transduced cell genomes [31] [91]. |
| Flow Cytometer | Multi-parameter analysis of transduction efficiency (%) and cell phenotype [31] [90]. | Determining the percentage of GFP+ CD3+ T-cells post-transduction [90]. |
| ELISA Kits | Quantification of secreted transgene protein concentrations in serum or supernatant [91]. | Monitoring Factor IX levels in mouse plasma after liver-directed gene therapy [91]. |
| Transduction Enhancers (e.g., Proteasome Inhibitors, IFNAR1 blockade) | Increase transduction efficiency by modulating host cell pathways [91]. | Pre-treatment in vivo to boost lentiviral transduction of hepatocytes [91]. |
The choice between viral and non-viral vectors remains a trade-off dominated by the inverse relationship between efficiency and safety. Viral vectors, particularly LV and AAV, currently deliver superior transduction rates and long-lasting expression, making them indispensable for diseases requiring permanent correction. However, their immunogenicity and genotoxicity risks are non-trivial. Non-viral vectors, led by LNPs and GalNAc conjugates, offer a safer, more scalable, and re-dosable alternative, albeit often with less potent or durable expression. The future of gene delivery lies in engineered solutions that mitigate these trade-offs—such as novel capsids for improved viral tropism and hybrid systems that combine the safety of non-viral platforms with the sustained efficacy of viral elements. For researchers, the decision must be guided by the specific therapeutic context: the target tissue, the required duration of expression, and the patient's underlying disease and immune status.
A critical challenge in modern gene therapy and genome editing is the safe and efficient delivery of genetic cargo into target cells. The choice of delivery system directly influences the therapeutic efficacy, specificity, and safety of the treatment. This guide provides an objective comparison of viral and non-viral delivery methods, with a focused analysis on their cargo capacity and versatility for transporting different forms of CRISPR-Cas9 editors.
The "cargo" in genome editing refers to the active biological components that perform the genetic modification, while the "vehicle" is the method used to transport these components into the cell [92]. For the CRISPR-Cas9 system, the cargo can be delivered in three primary forms, each with distinct implications for editing efficiency, timing, and safety [93] [94] [92].
1. Plasmid DNA (pDNA): A DNA plasmid encodes both the Cas9 protein and the guide RNA (gRNA). While stable and cost-effective to produce, it requires nuclear entry and subsequent transcription and translation. This delays editing activity to 24-48 hours and raises risks of random genomic integration and prolonged Cas9 expression, potentially increasing off-target effects [94] [92].
2. Messenger RNA (mRNA) and gRNA: This approach involves delivering mRNA that encodes the Cas9 protein, along with a separate gRNA. It leads to faster expression than pDNA as it bypasses the transcription step. It also reduces the risk of insertional mutagenesis and, due to its transient nature, can lower off-target effects. However, mRNA is inherently unstable and susceptible to degradation by nucleases [94] [92].
3. Ribonucleoprotein (RNP): This consists of the pre-assembled complex of the purified Cas9 protein and gRNA. RNP delivery is the most rapid, with gene editing detected within just one hour of delivery. It offers the highest specificity, minimal off-target effects due to its short cellular presence, and eliminates the risk of insertional mutagenesis. The key challenge is delivering the large, negatively charged protein complex across the cell membrane [94] [95] [92].
The following workflow outlines the journey of these different cargo types from delivery to genetic edit.
Comparative Analysis of Delivery Systems
Delivery vehicles are broadly categorized into viral, non-viral, and physical methods. Each system possesses inherent strengths and weaknesses that determine its suitability for specific cargo types and therapeutic applications.
Viral Delivery Vectors
Viral vectors are engineered viruses that have been modified to deliver genetic material into cells without causing disease.
- Adeno-Associated Viruses (AAVs): AAVs are small, non-pathogenic viruses with a high safety profile and low immunogenicity, making them one of the most common vectors for in vivo gene therapy [93] [96]. However, their severely limited packaging capacity of approximately 4.7 kb is a major constraint [93] [97] [96]. Since the commonly used Streptococcus pyogenes Cas9 (SpCas9) is about 4.2 kb, it is impossible to package it alongside a gRNA and promoter into a single AAV. Strategies to overcome this include using two separate AAVs or employing smaller, engineered Cas9 variants (e.g., Cas12a) [93].
- Adenoviral Vectors (AdVs): AdVs have a significantly larger cargo capacity of up to 36 kb, easily accommodating Cas9, multiple gRNAs, and donor DNA templates. They do not integrate into the host genome, ensuring transient expression. A key drawback is their tendency to provoke strong immune and inflammatory responses, which can be detrimental in therapeutic settings [93].
- Lentiviral Vectors (LVs): LVs can package large genetic payloads and integrate into the host genome, leading to long-term, stable expression. This is advantageous for ex vivo applications, such as engineering CAR-T cells. However, the integrating nature poses a risk of insertional mutagenesis, potentially activating oncogenes or disrupting tumor suppressor genes [93] [97].
Non-Viral and Physical Delivery Vectors
Non-viral methods use synthetic or biological materials to deliver cargo, while physical methods use physical force to transiently disrupt the cell membrane.
- Lipid Nanoparticles (LNPs): LNPs are synthetic particles that encapsulate nucleic acids or proteins, protecting them from degradation. They were successfully validated in mRNA COVID-19 vaccines and are now a leading platform for delivering CRISPR components like mRNA and RNP [93] [92]. A key challenge is enabling endosomal escape to prevent cargo degradation [93]. Recent advances like Selective Organ Targeting (SORT) LNPs allow for more precise tissue targeting [93].
- Extracellular Vesicles (EVs): EVs are natural, cell-derived lipid nanoparticles involved in intercellular communication. They offer high biocompatibility, low immunogenicity, and a natural ability to cross biological barriers [95] [98]. Recent innovative strategies, such as fusing RNA-binding proteins (e.g., MS2 coat protein) to EV membrane proteins (e.g., CD63), allow for efficient loading of Cas9 RNP complexes [95]. Engineered systems like ARMMs (ARRDC1-mediated microvesicles) have also demonstrated high editing efficiency in neuronal cells [98].
- Polymeric Nanoparticles: Cationic polymers, such as polyethyleneimine (PEI), can condense negatively charged cargoes into nanoparticles. While effective, high molecular weight PEI (e.g., 25K) is associated with cytotoxicity. Recent research focuses on linear or lower molecular weight PEI and chemical modifications to reduce toxicity while maintaining efficiency [99].
- Electroporation: This physical method uses electrical pulses to create temporary pores in the cell membrane, allowing cargo to enter the cytoplasm directly. It is highly efficient for ex vivo delivery of all cargo types, especially RNP, into sensitive cells like hematopoietic stem cells. This is the method used in the FDA-approved therapy CASGEVY [92]. A main limitation is reduced cell viability if conditions are not optimized [92] [96].
The table below summarizes the key characteristics of these delivery systems, providing a direct comparison of their cargo preferences and performance.
Table 1: Comparison of Delivery Systems for CRISPR-Cas9 Components
| Delivery System | Cargo Compatibility | Payload Capacity | Key Advantages | Major Limitations | Reported Editing Efficiency (Examples) |
|---|---|---|---|---|---|
| Adeno-Associated Virus (AAV) | DNA, ssRNA [93] | ~4.7 kb [93] [97] | Low immunogenicity, high tissue tropism, long-term expression [93] | Very limited capacity, pre-existing immunity in population [93] | Varies by serotype and target [93] |
| Adenovirus (AdV) | DNA [93] | Up to ~36 kb [93] | High capacity, infects dividing & non-dividing cells [93] | Strong immune response, potential toxicity [93] | Varies by serotype and target [93] |
| Lentivirus (LV) | RNA (converts to DNA) [93] | ~8 kb [93] | High capacity, stable genomic integration, broad tropism [93] | Risk of insertional mutagenesis, HIV backbone safety concerns [93] | Varies by pseudotype and target [93] |
| Lipid Nanoparticles (LNPs) | mRNA, RNP, siRNA [93] [92] | High & Flexible [93] | Rapid development, scalable production, low immunogenicity [93] [99] | Endosomal entrapment, potential for inflammation [93] [95] | >70% (Cas9 RNP in liver) [92], >97% gene knockdown (mRNA in hepatocytes) [99] |
| Extracellular Vesicles (EVs) | Protein, RNP, RNA [95] [98] | Moderate (Limited by EV size) [93] | Innate biocompatibility, natural targeting, cross biological barriers [95] [98] | Complex manufacturing, low native loading efficiency [93] [95] | High editing shown in neuronal APP gene [98] |
| Electroporation | RNP, mRNA, pDNA [92] [96] | High & Flexible [96] | Highly efficient for ex vivo delivery, direct cytosolic delivery [92] [96] | High stress on cells, reduced viability, mostly ex vivo [92] [96] | Up to 90% indels (CASGEVY, ex vivo RNP) [92] |
| Cationic Polymers (e.g., PEI) | pDNA, RNP [99] | High [99] | High condensation ability, promotes endosomal escape [99] | Cytotoxicity (especially high M.W. PEI) [99] | >90% transfection efficiency (in SKOV-3 cells) [99] |
Detailed Experimental Protocols
To illustrate how these delivery systems are evaluated in a research setting, here are detailed methodologies for two key approaches: EV-mediated RNP delivery and LNP-mediated mRNA delivery.
Protocol 1: EV-Mediated Cas9 RNP Delivery via Aptamer-Loading
This protocol, based on a 2025 Nature Communications study, details a modular system for loading Cas9 RNP into extracellular vesicles using high-affinity RNA aptamers [95].
- Objective: To achieve efficient loading and functional delivery of Cas9 RNP into target cells using engineered EVs.
- Materials:
- Plasmids: MS2 coat protein (MCP)-CD63 fusion, Cas9 (or variant), MS2-sgRNA expression vector.
- Cell Line: HEK293T cells (for EV production).
- Reagents: Transfection reagent, Tangential Flow Filtration (TFF) system, Size Exclusion Chromatography (SEC) columns, OptiPrep density gradient.
- Buffers: PBS, lysis buffer for Western blot.
- Methodology:
- Co-transfection: HEK293T cells are co-transfected with three plasmids: one expressing the MCP-CD63 fusion protein (the loading mechanism), one expressing Cas9, and one expressing the sgRNA engineered to contain MS2 RNA aptamers in its tetraloop and stemloop 2.
- EV Biogenesis and Loading: Inside the producer cell, the Cas9 protein and MS2-sgRNA form an RNP complex. The MS2 aptamers on the sgRNA are bound by the MCP domains displayed on the inner surface of budding EVs (e.g., on CD63 in multivesicular bodies), leading to efficient cargo loading during EV biogenesis.
- EV Isolation and Purification: At 48 hours post-transfection, the cell culture supernatant is collected. EVs are isolated and concentrated using Tangential Flow Filtration (TFF), followed by purification via Size Exclusion Chromatography (SEC) to remove non-vesicular contaminants.
- Characterization: Isolated EVs are characterized by:
- Nanoparticle Tracking Analysis (NTA): To determine particle size and concentration.
- Western Blot: To confirm the presence of EV markers (CD63, ALIX, TSG101) and Cas9, and the absence of contaminants like Calnexin.
- Density Gradient Centrifugation: To confirm Cas9 co-fractionates with EV markers.
- Functional Assay: The purified EVs are applied to recipient cells. Gene editing efficiency is quantified using methods like T7 Endonuclease I assay, Sanger sequencing followed by inference of CRISPR Edits (ICE), or next-generation sequencing (NGS) to calculate indel percentages.
The diagram below visualizes this modular loading strategy.
Protocol 2: LNP-Mediated Cas9 mRNA Delivery
This protocol outlines the key steps for formulating and testing LNPs encapsulating Cas9 mRNA and sgRNA for in vivo delivery [93] [92] [99].
- Objective: To formulate and functionally validate LNPs for the co-delivery of Cas9 mRNA and sgRNA to achieve in vivo genome editing.
- Materials:
- Nucleic Acids: Cas9 mRNA, sgRNA.
- Lipids: Ionizable cationic lipid, phospholipid, cholesterol, PEG-lipid.
- Equipment: Microfluidics device for LNP formation, NTA or DLS for size measurement.
- Animal Model: Mice (e.g., for liver targeting via intravenous injection).
- Methodology:
- LNP Formulation: Cas9 mRNA and sgRNA are mixed in an aqueous buffer at a defined ratio. This aqueous solution is combined with lipids dissolved in ethanol using a microfluidic device. The rapid mixing leads to the self-assembly of LNPs that encapsulate the nucleic acids, driven by electrostatic interactions between the ionizable lipids and the RNA backbone.
- LNP Characterization and Purification: The formulated LNPs are dialyzed or purified via tangential flow filtration (TFF) to remove ethanol and non-encapsulated RNA. They are then characterized for:
- Size and Polydispersity: Using Dynamic Light Scattering (DLS).
- Zeta Potential: Using DLS.
- Encapsulation Efficiency: Quantified using a Ribogreen assay.
- In Vivo Delivery: LNPs are administered systemically (e.g., via tail-vein injection) into mice. For liver targeting, the LNPs' surface properties can be modulated, for example, by incorporating apolipoprotein E (ApoE) ligands to enhance hepatocyte uptake [99].
- Efficiency Analysis: After a set period (e.g., 3-7 days), target tissues are harvested. Editing efficiency is analyzed by:
- NGS: of the target genomic locus to quantify the percentage of indel mutations.
- Western Blot or Immunohistochemistry: To assess changes in target protein expression.
The Scientist's Toolkit: Key Research Reagents
Successful execution of delivery experiments relies on a suite of specialized reagents and instruments.
Table 2: Essential Research Reagents and Tools
| Item | Function/Description | Example Use Case |
|---|---|---|
| Ionizable Cationic Lipids | Core component of LNPs; binds nucleic acids, promotes endosomal escape [93] [99]. | Formulating LNPs for mRNA/RNP delivery. |
| MS2-MCP System | High-affinity RNA-protein pair for loading cargo into engineered EVs [95]. | Modular loading of Cas9 RNP into EVs. |
| PEI (Polyethyleneimine) | Cationic polymer for condensing nucleic acids; "gold standard" for in vitro transfection but can be cytotoxic [99]. | In vitro transfection of plasmid DNA. |
| Tangential Flow Filtration (TFF) | A filtration method for concentrating and purifying nanoparticles like EVs and LNPs [95]. | Concentrating EV preparations from large volumes of cell culture supernatant. |
| Size Exclusion Chromatography (SEC) | Purifies nanoparticles based on size; removes protein aggregates and contaminants [95]. | Final polishing step for EV purification. |
| Nanoparticle Tracking Analysis (NTA) | Instrument that measures the size and concentration of particles in a suspension by tracking their Brownian motion [95]. | Characterizing the size distribution of isolated EVs or LNPs. |
| Electroporator | Device that delivers controlled electrical pulses to temporarily permeabilize cell membranes [92] [96]. | Ex vivo delivery of RNP into primary T cells or stem cells. |
The selection of a delivery system is a trade-off between cargo capacity, efficiency, safety, and practical application. Viral vectors like AAVs and LVs offer high transduction efficiency but are constrained by immunogenicity and, in the case of AAV, very limited cargo capacity. Non-viral vectors, particularly LNPs and engineered EVs, offer greater flexibility, improved safety profiles, and the ability to deliver sensitive cargoes like RNP complexes, which minimize off-target effects. Physical methods like electroporation remain the gold standard for many ex vivo applications.
The future of CRISPR delivery lies in the continued refinement of these non-viral platforms—improving their targeting capabilities, enhancing endosomal escape, and streamlining manufacturing. As the field progresses, the synergy between advanced cargo engineering (e.g., compact base editors) and sophisticated delivery vehicles will be paramount in realizing the full therapeutic potential of in vivo genome editing.
The selection of a delivery vector is a pivotal decision in gene therapy development, with significant implications for manufacturing, cost, and ultimate clinical success. The two primary approaches—viral vector-based methods (e.g., Lentivirus (LV), Adenovirus (Ad), Adeno-associated virus (AAV)) and non-viral methods (e.g., lipid nanoparticles (LNP), electroporation, sperm-mediated gene transfer (SMGT))—present distinct trade-offs [1]. Viral vectors are renowned for their high transduction efficiency, while non-viral systems offer advantages in safety, payload capacity, and simplified manufacturing scalability [1] [100]. This guide provides an objective, data-driven comparison of these platforms, focusing on the critical production metrics and commercial viability essential for researchers and drug development professionals.
Quantitative Comparison of Manufacturing Profiles
The manufacturing landscape for viral and non-viral vectors is evolving rapidly to meet growing clinical demand. The global viral vector, non-viral vector, and gene therapy manufacturing market is projected to grow from USD 0.70 billion to USD 2.3 billion by 2035, at a CAGR of 11.44% [101]. The broader viral vector gene therapy market specifically is forecast to grow from US$13.14 billion in 2024 to US$38.39 billion by 2034 [102]. The table below summarizes key comparative metrics.
Table 1: Direct Comparison of Viral vs. Non-Viral Vector Manufacturing and Commercialization
| Metric | Viral Vectors (e.g., AAV, LV) | Non-Viral Vectors (e.g., LNP, SMGT) |
|---|---|---|
| Typical Production Workflow | Complex, multi-step (cell culture, transfection, viral harvest, purification) [103] | Simpler, more scalable (chemical synthesis, nanoparticle formulation) [100] [103] |
| Production Cycle Timeline | Lengthy (can exceed nine months) [103] | Shorter production cycle [100] |
| Cost of Goods (COGs) | High; cited as a major barrier [103] | Can lower cost of goods by up to 60% [103] |
| Payload Capacity | Limited (AAV: ~4.7 kb; LV: ~8 kb) [1] | Larger payload capacity, suitable for polygenic indications [103] |
| Key Manufacturing Challenge | Constrained supply of GMP-grade vectors; meets only one-quarter of 2025 projected demand [103] | Optimizing lipid compositions for specific tissue targeting and transfection efficiency [104] |
| Scalability | Challenging due to complexity; requires sophisticated equipment and skilled personnel [102] | Highly scalable with standard pharmaceutical supply chains [103] |
| Commercial Readiness (Approved Therapies) | Dominant platform; 29 viral vector-based therapies approved as of 2025 [1] | Emerging; 6 non-viral vector-based therapies approved as of 2025 [1] |
| Market Share (2024) | AAV vectors held 38.54% of the gene therapy market share [103] | Non-viral LNP systems are the fastest-rising alternative, forecast to post a 24.34% CAGR through 2030 [103] |
Experimental Data and Protocol Analysis
Case Study: Non-Viral Lipid Nanoparticle (LNP) for Diabetes Gene Therapy
A 2025 preclinical study by Genprex collaborators provides a direct experimental model for non-viral manufacturing and application [104].
- Objective: To evaluate the transfection efficiency of nine different LNPs, prepared with patented LipexSil lipids, for delivering a GFP mRNA payload to pancreatic cells as a potential therapy for diabetes [104].
- Methodology:
- LNP Formulation: Nine distinct LNPs were synthesized using LipexSil lipids and loaded with GFP mRNA or Luciferase mRNA.
- In Vitro Transfection: Isolated mouse Islets of Langerhans were treated with the LNP formulations to assess transfection efficiency of α- and β-cells.
- In Vivo Delivery and Specificity: The selected LNP (ALX-184 with Luciferase mRNA) was administered via infusion into the mouse common bile duct to target the pancreas. Luciferase activity was measured in the pancreas and other organs to determine targeting specificity [104].
- Key Outcomes:
- Two specific LNPs demonstrated high efficiency in transfecting pancreatic α- and β-cells in vitro.
- The ALX-184 LNP showed exceptional in vivo targeting, with luciferase activity in the pancreas 100 times greater than in other organs.
- The selected LNP was less toxic than a commercially used lipid and successfully crossed the pancreatic duct's basement membrane to transfect target cells [104].
Case Study: Sperm-Mediated Gene Transfer (SMGT) in Swine
SMGT represents a simple and cost-effective non-viral method for creating transgenic animals, with proven scalability in large species [105].
- Objective: To investigate the influence of SMGT treatment and exogenous DNA uptake on sperm quality parameters and in vitro fertilization (IVF) ability over an extended period [105].
- Methodology:
- Sperm Treatment: Boar spermatozoa were washed to remove seminal plasma and then coincubated with exogenous DNA (at standard 5 μg/mL and high 100 μg/mL concentrations) for up to 48 hours.
- Quality Assessment: Sperm were analyzed at multiple time points for objectively measured motility (OM, PM), viability (V), mitochondrial membrane potential (MMP), and acrosome damage (AD).
- Functional Assay: IVF trials were performed using SMGT-treated spermatozoa stored for 24 hours to assess cleavage and developmental rates [105].
- Key Outcomes:
- The SMGT protocol and DNA binding did not significantly impair sperm quality parameters (motility, viability, MMP) for at least 24 hours.
- Even with a 20-fold larger amount of DNA (100 μg/mL), sperm quality was not significantly affected.
- In IVF trials 24 hours post-DNA uptake, SMGT-treated sperm maintained good functionality, with cleavage rates of 60% (vs. 58% control) and developmental rates of 41% (vs. 48% control) [105].
Visualizing Manufacturing Workflows
The following diagrams illustrate the core operational workflows for producing viral and non-viral vectors, highlighting the complexity and key stages involved.
The Scientist's Toolkit: Key Research Reagents
Successful vector development and manufacturing rely on a suite of specialized reagents and materials. The table below details essential components for both viral and non-viral platforms.
Table 2: Essential Research Reagents for Vector Development and Manufacturing
| Reagent/Material | Function in Manufacturing/Research | Platform Applicability |
|---|---|---|
| LipexSil Lipids | Patented lipids used to formulate lipid nanoparticles (LNPs) for efficient mRNA delivery and reduced toxicity [104] | Non-Viral (LNP) |
| Plasmid DNA (pDNA) | Backbone for producing viral vectors or as a genetic payload for non-viral methods; a critical raw material [103] | Viral & Non-Viral |
| Cap Analogs | Modified nucleotides (e.g., CleanCap) used in IVT to create a 5' cap structure on mRNA, enhancing stability and translation [100] | Non-Viral (mRNA) |
| T7 RNA Polymerase | Enzyme for in vitro transcription (IVT) of mRNA from a DNA template [100] | Non-Viral (mRNA) |
| Modified Nucleotides | Nucleotides (e.g., pseudouridine-Ψ) that replace standard ones in IVT mRNA to reduce immunogenicity and enhance stability [100] | Non-Viral (mRNA) |
| Cell Lines (HEK293) | Immortalized cell lines used as factories to produce viral vectors like AAV and LV [1] | Viral |
| Chromatography Resins | Resins (e.g., affinity, ion-exchange) for purifying viral vectors from cell culture lysates, a key bottleneck [101] | Viral |
| Sperm Media Extender (e.g., SFM) | Specialized buffer for washing and preserving spermatozoa during SMGT procedures, maintaining fertility [105] | Non-Viral (SMGT) |
The choice between viral and non-viral vector manufacturing strategies involves a direct trade-off between transduction efficiency and scalability. Viral vectors, particularly AAV and LV, currently dominate the clinical landscape but face significant challenges related to manufacturing complexity, high costs, and constrained supply chains [1] [103]. In contrast, non-viral platforms, including LNP and SMGT, offer compelling advantages in manufacturing simplicity, reduced cost of goods, and a superior safety profile, which facilitates re-dosing [104] [103].
The market is poised for evolution. While AAV vectors currently lead in market share, non-viral systems are projected to be the fastest-growing segment [103]. Future growth will be driven by technological advancements in lipid chemistry, continuous manufacturing processes, and AI-driven optimization, which will collectively address current limitations and expand the therapeutic reach of gene therapies to a broader patient population [102] [103].
The selection of an appropriate gene delivery vector is a foundational decision that directly determines the success of gene therapy development. This choice is guided by a complex matrix of factors, including the therapeutic indication, target cell type, required duration of transgene expression, and the biological properties of the vector itself [38] [1]. For decades, the field operated on a relatively simple paradigm: adeno-associated viruses (AAV) for in vivo gene delivery and lentiviruses (LV) for ex vivo cell engineering [38]. However, the rapid advancement of gene editing technologies and the clinical validation of non-viral delivery systems are fundamentally reshaping this landscape.
The emergence of CRISPR-based editing and the demand for delivering larger genetic payloads have exposed the limitations of traditional viral capsids [38]. Simultaneously, the manufacturing success of lipid nanoparticles (LNPs) during the COVID-19 pandemic has demonstrated the viability of non-viral vectors as scalable and re-dosable alternatives [38] [3]. Within the specific context of Sperm-Mediated Gene Transfer (SMGT), this evolution is particularly relevant, as researchers seek efficient and safe methods to generate genetically modified animal models [106] [107]. This guide provides a technical, data-driven framework to navigate this critical decision, equipping researchers with the comparative data and methodologies needed to select the optimal vector for their therapeutic indication and target cell.
Comparative Vector Analysis: Technical Specifications and Applications
The choice between viral and non-viral vectors involves trade-offs between cargo capacity, persistence of expression, immunogenicity, and manufacturing complexity. The tables below summarize the core characteristics and application landscapes of the primary vector systems.
Table 1: Technical and Biological Comparison of Major Gene Delivery Vectors
| Feature | AAV (Viral) | Lentivirus (Viral) | LNP (Non-Viral) |
|---|---|---|---|
| Primary Use Case | In vivo gene replacement for static tissues (CNS, eye, liver) [38] | Ex vivo cell therapy (CAR-T, HSCs) [38] [1] | Gene editing (CRISPR/mRNA), vaccines [38] |
| Cargo Capacity | ~4.7 kb (strict limit) [38] [108] | ~10 kb (moderate) [38] | Flexible / High (virtually unlimited) [38] |
| Genetic Persistence | Episomal (long-term in non-dividing cells) [38] [22] | Integrated (permanent in dividing cells) [38] [22] | Transient (ideal for editing) [38] |
| Immunogenicity | High (pre-existing NAbs exclude patients, prevents re-dosing) [38] | Low (mostly used ex vivo) [38] | Low (re-dosable, though PEG-antibodies are a consideration) [38] |
| Key Manufacturing Bottleneck | Empty/Full capsid separation [38] | Viral stability & titer (shear-sensitive) [38] | Lipid purity & microfluidic fouling [38] |
| Tropism / Targeting | Determined by capsid serotype (e.g., AAV9 for CNS) [38] | Broad tropism, can be pseudotyped (e.g., with VSV-G) [1] | Naturally accumulates in liver; targeting other tissues requires complex lipid engineering [38] |
Table 2: Approved Therapies and Clinical Applications by Vector Type
| Vector Type | Example Approved Therapies | Therapeutic Area | Administration Route |
|---|---|---|---|
| Adeno-associated Virus (AAV) | Luxturna (inherited retinal dystrophy), Zolgensma (spinal muscular atrophy), Hemgenix (hemophilia B) [1] [3] | Monogenic inherited diseases (ophthalmic, neuromuscular, metabolic) [1] | In vivo (subretinal, intravenous) [1] |
| Lentivirus (LV) | Zynteglo (beta-thalassemia), Libmeldy (metachromatic leukodystrophy), multiple CAR-T therapies (e.g., Kymriah) [1] [3] | Hematological disorders, ex vivo cell engineering for oncology [1] | Ex vivo (patient cells modified and reinfused) [1] |
| Lipid Nanoparticle (LNP) | Onpattro (hATTR amyloidosis), COVID-19 mRNA vaccines [1] [3] | Protein replacement, gene silencing (RNAi), vaccines [3] | In vivo (intravenous, intramuscular) [1] |
| N-acetylgalactosamine (GalNAc) | Givlaari (acute hepatic porphyria), Oxlumo (primary hyperoxaluria type 1) [1] [3] | Liver-targeted RNA therapies [3] | In vivo (subcutaneous) [1] |
Experimental Protocols for Vector Evaluation
Protocol: Optimizing Sperm-Mediated Gene Editing (SMGE) with Methyl-β-Cyclodextrin
The MBCD-SMGE technique demonstrates how non-viral methods can be adapted for efficient gene editing in a specialized application, relevant for creating animal models [107].
Objective: To enhance the uptake of the CRISPR/Cas9 system into sperm cells for the generation of targeted mutant blastocysts and mice [107]. Key Reagents:
- Spermatozoa: Collected from B6D2F1 mouse strain [107].
- Gene Construct: pCAG-eCas9-GFP-U6-gRNA plasmid [107].
- Chemical Agent: Methyl-β-cyclodextrin (MBCD) in c-TYH medium [107].
- Oocytes: Collected from superovulated CD1 female mice [107].
Methodology:
- Sperm Treatment: Incubate sperm in c-TYH medium supplemented with varying concentrations of MBCD (0, 0.75, 1, and 2 mM) in the presence of the pgRNA-Cas9 plasmid (20 ng/μl) for 30 minutes [107].
- Functional Assessment: Evaluate sperm motility, and acrosomal reaction. Quantify extracellular reactive oxygen species (ROS) and the copy number of internalized plasmid per sperm cell [107].
- In Vitro Fertilization (IVF): Perform IVF using treated sperm and collected oocytes. Culture the resulting zygotes in mKSOM medium [107].
- Embryo Analysis: Assess fertilization rate, early embryonic development, and the rate of GFP-positive blastocysts (indicating successful transfection) [107].
- Genotype Validation: Sequence the target locus (e.g., Gdf8 gene) in blastocysts and offspring to validate targeted indels [107].
Conclusion: This protocol highlights that cholesterol removal from the sperm membrane via MBCD increases plasmid uptake and enhances the production of transfected embryos, providing a efficient non-viral method for generating targeted mutant models [107].
Protocol: Assessing Non-Viral Vector Transfection in the Retinal Pigment Epithelium (RPE)
This methodology is critical for evaluating non-viral vectors for ocular gene therapy, an area where AAV's cargo capacity can be limiting [108].
Objective: To test the transfection efficiency and safety of polymer-compacted DNA nanoparticles in the retinal pigment epithelium (RPE) [108]. Key Reagents:
- Vector: Plasmid DNA compacted with specialty carriers like polylysine (e.g., CK30) or polyethyleneimine (PEI) [108].
- Animal Model: Wild-type or genetically defective mice (e.g., rpe65-/- or lrat-/- for disease models) [108].
- Control: AAV vectors carrying a reporter or therapeutic gene (e.g., RPE65) [108].
Methodology:
- Vector Preparation: Compact plasmid DNA containing a therapeutic gene (e.g., RPE65) and a reporter gene with the chosen cationic polymer to form stable, nearly charge-neutral nanoparticles [108].
- Subretinal Injection: Anesthetize the mouse and perform a subretinal injection to deliver the nanoparticle suspension directly into the subretinal space, ensuring contact with the RPE layer [108]. Intravitreal injection can be used as a less efficient control route [108].
- In Vivo Assessment: Monitor transgene expression over time using in vivo imaging (e.g., fluorescence from a reporter protein). Assess functional rescue in disease models by electroretinography (ERG) [108].
- Histological Analysis: Process retinal tissue for immunohistochemistry to confirm RPE-specific protein expression and examine tissue morphology and potential cytotoxicity [108].
- Immunogenicity Evaluation: Analyze humoral and cellular immune responses against the vector and transgene compared to AAV-injected controls [108].
Conclusion: This protocol allows for the direct comparison of non-viral and viral vectors in a therapeutically relevant tissue, evaluating key parameters such as transfection efficiency, duration of expression, and safety profile [108].
Visualization of Workflows and Pathways
Decision Framework for Vector Selection
The following diagram outlines a logical decision pathway for selecting the most appropriate gene delivery vector based on key experimental and therapeutic requirements.
MBCD-SMGE Experimental Workflow
This flowchart details the key steps in the Methyl-β-Cyclodextrin Sperm-Mediated Gene Editing protocol, an advanced non-viral method for generating genetically modified models.
The Scientist's Toolkit: Essential Research Reagents
Successful vector evaluation and development rely on a suite of specialized reagents and tools. The following table catalogues key solutions used in the featured experiments and the broader field.
Table 3: Essential Research Reagents for Vector Development and Evaluation
| Reagent / Solution | Function / Application | Example Use Case |
|---|---|---|
| Methyl-β-Cyclodextrin (MBCD) | A cyclic oligosaccharide that removes cholesterol from cell membranes, enhancing permeability and exogenous DNA uptake [107]. | Optimizing sperm-mediated gene transfer (SMGT) by increasing CRISPR/Cas9 plasmid uptake into sperm cells [107]. |
| Cationic Polymers (PEI, CK30 Polylysine) | Positively charged polymers that compact negatively charged DNA into nanoparticles, protecting it and facilitating cellular uptake [108]. | Forming stable, nearly charge-neutral DNA nanoparticles for efficient in vivo gene delivery to tissues like the retina [108]. |
| Lipid Nanoparticles (LNPs) | Synthetic, scalable vesicles that encapsulate and deliver nucleic acids (mRNA, siRNA, DNA) into cells via endocytosis [38] [3]. | Delivering CRISPR/Cas9 components for gene editing; platform for mRNA vaccines and therapeutics [38] [3]. |
| Adeno-Associated Virus (AAV) Serotypes | Engineered viral capsids with distinct tissue tropism (e.g., AAV9 for CNS, AAV8 for liver) used for in vivo gene delivery [38] [1]. | Gene replacement therapy in post-mitotic tissues for diseases like spinal muscular atrophy (Zolgensma) [1]. |
| Lentiviral Vectors (VSV-G pseudotyped) | Retroviral vectors capable of infecting both dividing and non-dividing cells and integrating their payload into the host genome [38] [1]. | Engineering patient-derived T-cells with Chimeric Antigen Receptors (CAR) for cancer immunotherapy [1] [3]. |
| Specialized Cell Culture Media (c-TYH, mKSOM) | Chemically defined media optimized for specific cellular processes, such as sperm capacitation (c-TYH) and embryo culture (mKSOM) [107]. | Supporting in vitro fertilization and early embryonic development following sperm-mediated gene editing procedures [107]. |
The decision framework for selecting a gene therapy vector is no longer governed by rigid rules but is instead a nuanced process tailored to specific therapeutic needs. The prevailing trend is towards pragmatism and delivery agnosticism, where the biological requirements of the therapy dictate the tool, rather than historical precedent [38]. AAV remains powerful for durable in vivo expression in accessible tissues, lentiviruses are entrenched for ex vivo cell engineering, and LNPs have emerged as the premier vehicle for transient expression, particularly for gene editing [38] [3].
Future advancements will continue to blur the lines between viral and non-viral systems. We are already seeing the rise of hybrid approaches, such as virus-like particles (VLPs) that mimic viral entry without carrying viral genetic material, and the refinement of exosomes as biomimetic delivery vehicles [38]. Furthermore, intensive vector engineering—including capsid engineering to evade immune responses and alter tropism, and the optimization of lipid chemistries to direct LNPs beyond the liver—will expand the reach of both platforms [38] [22]. For researchers, the most successful strategy will be to maintain a flexible toolkit, selecting the optimal vector through a clear-eyed assessment of its capabilities against the stringent demands of the therapeutic indication and target cell.
Conclusion
The choice between viral and non-viral vectors is not a matter of superiority but of strategic alignment with therapeutic goals. Viral vectors, particularly LV and AAV, currently offer high efficiency and durable expression, powering landmark therapies for rare diseases. Meanwhile, non-viral vectors, led by LNPs, provide a safer, more scalable profile with growing clinical validation. The future lies in continued innovation to overcome immunogenicity, enhance targeting, and streamline manufacturing. Convergence is likely, with hybrid approaches combining the precision of viral tropism with the safety of synthetic systems. For researchers and developers, a nuanced understanding of these platforms is paramount for designing the next generation of transformative gene therapies that are not only effective but also accessible.
References
- 1. Viral and non-viral vectors in gene therapy: current state and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12271757/]
- 2. Viral vector platforms within the gene therapy landscape [https://www.nature.com/articles/s41392-021-00487-6]
- 3. The State and Future of Viral, Nonviral Vectors for Gene ... [https://www.ajmc.com/view/the-state-and-future-of-viral-non-viral-vectors-for-gene-therapy]
- 4. Current clinical applications of AAV-mediated gene therapy [https://www.sciencedirect.com/science/article/pii/S152500162500365X]
- 5. Delivery Success in Gene Editing [https://www.aldevron.com/blog/lnp-delivery-technology]
- 6. Emerging Technologies Tackling Adeno-Associated ... [https://www.mdpi.com/1999-4923/17/11/1492]
- 7. Emerging Perspectives on Gene Therapy Delivery for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9788053/]
- 8. Delivery Systems: Gene . Viral - vs Vectors Non Viral [https://www.azolifesciences.com/article/Gene-Delivery-Systems-Viral-vs-Non-Viral-Vectors.aspx]
- 9. Exploring Delivery Methods for Gene Manufacturing Therapy [https://www.thermofisher.com/blog/behindthebench/unlocking-the-future-exploring-viral-and-non-viral-delivery-methods-for-gene-therapy-manufacturing/]
- 10. Choosing the Right Vector for Gene Therapy and Research [https://www.cd-genomics.com/biomedical-ngs/resource/aav-lentivirus-adenovirus-gene-therapy-vaccine.html]
- 11. Viral Vectors Compared: AAV vs. Lentivirus vs. Adenovirus [https://synapse.patsnap.com/article/viral-vectors-compared-aav-vs-lentivirus-vs-adenovirus]
- 12. Adeno-associated virus as a delivery vector for gene ... [https://www.nature.com/articles/s41392-024-01780-w]
- 13. AAV Vs. Lentiviral Vectors - Life in the Lab [https://www.thermofisher.com/blog/life-in-the-lab/aav-vs-lv/]
- 14. Viral and non-viral vectors in gene therapy: current state ... [https://www.sciencedirect.com/science/article/pii/S2352396425002786]
- 15. Emerging non-viral vectors for gene delivery [https://jnanobiotechnology.biomedcentral.com/articles/10.1186/s12951-023-02044-5]
- 16. siRNA Delivery: LNP vs. GalNAc vs. Polymers [https://www.bocsci.com/research-area/sirna-delivery-vehicle-comparison-lnp-vs-galnac-vs-polymers.html?srsltid=AfmBOoo8CLDNqial7SM6JJ_xXnWLWMr5eYWFh8nv4B0A6yneouM-Y_2P]
- 17. Circular single stranded DNA potentiates non-viral gene ... [https://www.nature.com/articles/s41467-025-66318-2]
- 18. Non-viral vectors for RNA delivery - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC8743282/]
- 19. GalNAc-Lipid nanoparticles enable non-LDLR dependent ... [https://www.nature.com/articles/s41467-023-37465-1]
- 20. Appraisal for the Potential of Viral and Nonviral Vectors in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9407213/]
- 21. Optimizing viral transduction in immune cell therapy ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06524-0]
- 22. Vectors in Gene Therapy: Viral & Non- ... [https://www.cellandgene.com/topic/cell-gene-therapy-vectors]
- 23. Molecular Engineering of Virus Tropism [https://www.mdpi.com/1422-0067/25/20/11094]
- 24. Viral and Non-Viral Systems to Deliver Gene Therapeutics ... [https://www.mdpi.com/1422-0067/25/13/7333]
- 25. Viral vs Non-Viral Vectors in Gene Therapy [https://www.phacilitate.com/insights-resources/gene-therapy-big-debate-viral-vectors-vs-non-viral-vectors]
- 26. Viral and Non-Viral Systems to Deliver Gene Therapeutics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11242246/]
- 27. A Review of CGT: Viral and Non-Viral Approaches [https://proventainternational.com/oncology-viral-and-non-viral-approaches-article/]
- 28. From the Editors: Cell & Gene Therapy Approvals in 2024 [https://www.isctglobal.org/telegrafthub/blogs/ken-ip1/2025/01/16/cell-gene-therapy-approvals-in-2024]
- 29. FDA Approves Gene Therapy for Treatment of Spinal ... [https://www.fda.gov/news-events/press-announcements/fda-approves-gene-therapy-treatment-spinal-muscular-atrophy]
- 30. Novartis receives FDA approval for Itvisma®, the only gene ... [https://www.novartis.com/news/media-releases/novartis-receives-fda-approval-itvisma-only-gene-replacement-therapy-children-two-years-and-older-teens-and-adults-spinal-muscular-atrophy-sma]
- 31. Optimizing viral transduction in immune cell therapy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12046913/]
- 32. Clinical hematopoietic stem cell-based gene therapy [https://www.sciencedirect.com/science/article/pii/S1525001625003089]
- 33. Current landscape of vector safety and genotoxicity after ... [https://www.nature.com/articles/s41375-025-02585-8]
- 34. From ex vivo to in vivo chimeric antigen T cells ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-024-06052-3]
- 35. Next generation targeted non-genotoxic conditioning for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12520940/]
- 36. Retroviral Vector Technology for Gene Therapy [https://www.sciencedirect.com/science/article/pii/S002228362500539X]
- 37. Rethinking lentiviral manufacturing for cell and gene ... [https://www.insights.bio/cell-and-gene-therapy-insights/journal/article/3577/rethinking-lentiviral-manufacturing-for-cell-and-gene-therapies-from-platform-design-to-pointofcare-delivery]
- 38. Viral vs. Non-Viral Gene Delivery: A Critical Choice for ... [https://www.drugdiscoverynews.com/viral-vs-non-viral-gene-delivery-a-critical-choice-for-gene-therapy-developers-16819]
- 39. AAV9-mediated MYBPC3 gene therapy with optimized ... [https://www.nature.com/articles/s41467-025-57481-7]
- 40. AAV-GAD gene therapy eases Parkinson's symptoms in ... [https://parkinsonsnewstoday.com/news/aav-gad-gene-therapy-eases-parkinsons-symptoms-early-study/]
- 41. Study suggests promising gene therapy for FOXG1 syndrome [https://www.buffalo.edu/news/releases/2024/06/study-suggests-promising-gene-therapy-for-FOXG1-syndrome.html]
- 42. The postnatal injection of AAV9-FOXG1 rescues corpus ... [https://pubmed.ncbi.nlm.nih.gov/39022742/]
- 43. Rett Syndrome Gene Therapy Study Shows Positive ... [https://www.packgene.com/news/021425-rett-syndrome/]
- 44. First participants randomized in AskBio Phase II gene ... [https://www.askbio.com/first-participants-randomized-in-askbio-phase-ii-gene-therapy-trial-for-parkinsons-disease/]
- 45. Investigational Gene Therapies for Parkinson's Disease - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12263715/]
- 46. RNA Therapeutics: Delivery Problems and Solutions—A Review [https://pmc.ncbi.nlm.nih.gov/articles/PMC12567017/]
- 47. The current and future landscape of RNA-based therapies ... [https://www.nature.com/articles/s43856-025-01166-1]
- 48. Beyond LNPs 4 Non-Viral Delivery Vehicles Expanding ... [https://www.advancingrna.com/doc/beyond-lnps-non-viral-delivery-vehicles-expanding-the-possibilities-of-mrna-therapeutics-0001]
- 49. mRNA Delivery Systems 2.0: Engineering Extrahepatic ... [https://www.sciencedirect.com/science/article/pii/S259000642501155X]
- 50. The impact of lipid compositions on siRNA and mRNA ... [https://www.sciencedirect.com/science/article/pii/S0928098725001812]
- 51. RNA Interference Drug Delivery Market Size 2025 to 2034 [https://www.precedenceresearch.com/rna-interference-drug-delivery-market]
- 52. Administration route governs the therapeutic efficacy ... [https://jnanobiotechnology.biomedcentral.com/articles/10.1186/s12951-021-00803-w]
- 53. Local and systemic biodistribution of a small-molecule ... [https://pubmed.ncbi.nlm.nih.gov/39774101/]
- 54. Evaluation of biodistribution by local versus systemic ... [https://pubmed.ncbi.nlm.nih.gov/17013741/]
- 55. The Effect of Injection Routes on the Biodistribution ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3725601/]
- 56. Impact of ionizable lipid type on the pharmacokinetics and ... [https://www.sciencedirect.com/science/article/pii/S0168365925005656]
- 57. Pharmacokinetics, biodistribution and cell uptake of ... [https://www.sciencedirect.com/science/article/pii/S0169409X15000101]
- 58. Immune Responses to Viral Gene Therapy Vectors - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7054714/]
- 59. Progress and Prospects: Immune to Responses - PMC Viral Vectors [https://pmc.ncbi.nlm.nih.gov/articles/PMC3044498/]
- 60. Adeno-associated viral vector-mediated immune responses [https://www.sciencedirect.com/science/article/pii/S0163725819302050]
- 61. Biomimetic artificial enveloped viral vectors: Overcoming ... [https://www.sciencedirect.com/science/article/abs/pii/S3050562325001345]
- 62. Stem cell gene therapy: the risks of insertional ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3508510/]
- 63. Chance or necessity? Insertional Mutagenesis in Gene ... [https://www.sciencedirect.com/science/article/pii/S1525001603003538]
- 64. Evaluating Risks of Insertional Mutagenesis by DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3602164/]
- 65. Advancing ex vivo gene therapy: the role of lentiviral ... [https://www.sciencedirect.com/science/article/pii/S1359644625002235]
- 66. Gene delivery systems explained. "Viral vs. Non ... [https://www.lqventures.com/gene-delivery-systems-explained-viral-vs-non-viral-vectors-debate/]
- 67. Non-Viral Gene Delivery Systems: Current Advances and ... [https://pubmed.ncbi.nlm.nih.gov/40874945/]
- 68. Progress and challenges in viral vector manufacturing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4802372/]
- 69. Review Challenges in scaling up AAV-based gene therapy ... [https://www.sciencedirect.com/science/article/pii/S0167779923001233]
- 70. Addressing the Challenges of Commercial-Scale ... [https://www.insights.bio/cell-and-gene-therapy-insights/journal/article/325/addressing-the-challenges-of-commercialscale-manufacture-of-viral-vectors-for-cart-therapies]
- 71. Non Viral Transfection Reagents Market Size, Share 2025- ... [https://www.coherentmi.com/industry-reports/non-viral-transfection-reagents-market]
- 72. Overview of the non-viral gene delivery methods [https://insidetx.com/resources/reviews/overview-of-the-non-viral-gene-delivery-methods/]
- 73. Transfection Technologies for Next-Generation Therapies [https://pubmed.ncbi.nlm.nih.gov/40807133/]
- 74. Non-Viral Transfection Methods Optimized for Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3689559/]
- 75. Enhancing non-viral gene delivery to human T cells ... [https://www.sciencedirect.com/science/article/pii/S0753332225000149]
- 76. Environmental parameters influence non-viral transfection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3307818/]
- 77. Enhancing non-viral DNA delivery systems [https://www.sciencedirect.com/science/article/pii/S0168365924008459]
- 78. Advances in AAV capsid engineering: Integrating rational ... [https://www.sciencedirect.com/science/article/pii/S1525001625002655]
- 79. Identification and validation of novel engineered AAV ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11348808/]
- 80. Design Strategies for Novel Lipid Nanoparticle for mRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12497691/]
- 81. Transformative Advances in Viral Vector Manufacturing [https://www.pharmasalmanac.com/articles/transformative-advances-in-viral-vector-manufacturing-unlocking-commercial-scalability-consistency-and-cost-effectiveness]
- 82. Modeling viral vector capacity: Healthy growth by 2031 [https://www.bioprocessintl.com/facilities-capacity/modeling-viral-vector-capacity-healthy-growth-by-2031]
- 83. Viral and non-viral vectors for gene therapy in the treatment ... [https://www.sciencedirect.com/science/article/abs/pii/S0098299725000640]
- 84. Past, Present, and Future of Viral Vector Vaccine Platforms [https://pmc.ncbi.nlm.nih.gov/articles/PMC12115715/]
- 85. Phase 1/2 safety and immunogenicity of IIBR-100 in ... [https://pubmed.ncbi.nlm.nih.gov/41106035/]
- 86. Phase 1/2 safety and immunogenicity of IIBR-100 in ... [https://www.sciencedirect.com/science/article/abs/pii/S0264410X2501134X]
- 87. Safety and immunogenicity of an mRNA-based RSV ... [https://pubmed.ncbi.nlm.nih.gov/40953212/]
- 88. Evaluation of AAV transduction efficiency via multiple ... [https://www.sciencedirect.com/science/article/pii/S0306452225002283]
- 89. Is the tide turning on viral vectors? The shifting landscape ... [https://www.bioprocessintl.com/therapeutic-class/is-the-tide-turning-on-viral-vectors-the-shifting-landscape-of-gene-therapy-delivery]
- 90. a novel transduction device for efficient and scalable gene ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06836-1]
- 91. Enhancing the potency of in vivo lentiviral vector mediated ... [https://www.nature.com/articles/s41467-025-60073-0]
- 92. CRISPR/Cas9 Delivery Systems to Enhance Gene Editing ... [https://www.mdpi.com/1422-0067/26/9/4420]
- 93. CRISPR Delivery Methods: Cargo, Vehicles, and Challenges [https://www.synthego.com/blog/delivery-crispr-cas9/]
- 94. Recent advances in the delivery and applications of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10052255/]
- 95. A modular strategy for extracellular vesicle-mediated ... [https://www.nature.com/articles/s41467-025-65995-3]
- 96. Challenges and Opportunities in the Application of CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12503735/]
- 97. Viral Vector vs CRISPR Resources [https://bioinnovatise.com/articles/viral-vector-vs-crispr/]
- 98. Engineering ARMMs for improved intracellular delivery of ... [https://www.sciencedirect.com/science/article/pii/S2773041725000186]
- 99. Discover the recent progress of nonviral delivery carriers ... [https://www.news-medical.net/whitepaper/20240110/Discover-the-Recent-Progress-of-Nonviral-Delivery-Carriers-for-CRISPRCas9-Systems.aspx]
- 100. Progress and prospects of mRNA-based drugs in pre- ... [https://www.nature.com/articles/s41392-024-02002-z]
- 101. Viral Vector Manufacturing, Non-Viral ... [https://www.businesswire.com/news/home/20250409404262/en/Viral-Vector-Manufacturing-Non-Viral-Vector-Manufacturing-and-Gene-Therapy-Manufacturing-Market-Research-2025-2035---ResearchAndMarkets.com]
- 102. Viral Vector Gene Therapy Market to Reach USD 38.39 ... [https://www.towardshealthcare.com/insights/viral-vector-gene-therapy-market-sizing]
- 103. Gene Therapy Market Size, Growth Analysis (2025-2030) [https://www.mordorintelligence.com/industry-reports/gene-therapy-market]
- 104. Genprex Collaborators Present Research on Non-Viral ... [https://www.genprex.com/2025/06/23/genprex-collaborators-present-research-on-non-viral-approach-to-diabetes-gene-therapy-using-lipid-nanoparticle-delivery-system-at-the-2025-american-diabetes-association-85th-scientific-sessi/]
- 105. Sperm-mediated gene transfer–treated spermatozoa ... [https://www.sciencedirect.com/science/article/abs/pii/S0093691X09003446]
- 106. Sheep and Goat Genome Engineering - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC6735269/]
- 107. Methyl β-Cyclodextrin-sperm-mediated gene editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10811837/]
- 108. A review of therapeutic prospects of non-viral gene therapy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3742069/]