Decoding Idiopathic POI: A Deep Dive into a 163-Gene NGS Panel for Researchers
Premature Ovarian Insufficiency (POI), affecting 1-3.7% of women, remains idiopathic in a significant proportion of cases, posing a major challenge in female infertility.
Decoding Idiopathic POI: A Deep Dive into a 163-Gene NGS Panel for Researchers
Abstract
Premature Ovarian Insufficiency (POI), affecting 1-3.7% of women, remains idiopathic in a significant proportion of cases, posing a major challenge in female infertility. This article explores the application of a custom next-generation sequencing (NGS) panel targeting 163 genes known or suspected in ovarian function. We review the substantial diagnostic yield of this approach, which can identify pathogenic variants in over 57% of idiopathic POI cases by uncovering defects across diverse biological pathways, including meiosis, DNA repair, and folliculogenesis. For researchers and drug development professionals, this analysis covers the panel's technical implementation, data interpretation challenges, and its pivotal role in validating novel gene-disease associations through large-scale cohort studies. The discussion extends to how these genetic insights are illuminating new therapeutic targets and paving the way for personalized medicine strategies in ovarian aging.
The Genetic Landscape of POI: From Idiopathic Mystery to Actionable Insights
Defining the Clinical and Genetic Heterogeneity of POI
Premature Ovarian Insufficiency (POI) is a clinically heterogeneous condition characterized by the loss of ovarian function before the age of 40, affecting approximately 1%-3.7% of women [1] [2]. The diagnosis requires the presence of menstrual disturbances (amenorrhea or oligomenorrhea for at least four months) and elevated serum follicle-stimulating hormone (FSH) levels (>25 U/L on two occasions at least four weeks apart) [1]. POI carries significant short-term and long-term health consequences, including infertility, vasomotor symptoms, increased risks of osteoporosis, cardiovascular disease, and cognitive decline [1]. The etiological landscape of POI has evolved substantially in recent decades, with a notable shift from predominantly idiopathic cases to an increased identification of genetic, autoimmune, and iatrogenic causes [1].
The Evolving Etiological Spectrum of POI
Contemporary research reveals a complex multifactorial etiology behind POI. A recent comparative study of historical (1978-2003) and contemporary (2017-2024) cohorts demonstrated a significant redistribution of causative factors, highlighted in Table 1 [1].
Table 1: Changing Etiological Distribution of POI Over Time
| Etiological Category | Historical Cohort (1978-2003) Prevalence (%) | Contemporary Cohort (2017-2024) Prevalence (%) | P-Value |
|---|---|---|---|
| Genetic | 11.6 | 9.9 | Not Significant |
| Autoimmune | 8.7 | 18.9 | <0.05 |
| Iatrogenic | 7.6 | 34.2 | <0.05 |
| Idiopathic | 72.1 | 36.9 | <0.05 |
This striking redistribution, with idiopathic cases halving from 72.1% to 36.9%, reflects advances in diagnostic capabilities and changing clinical profiles. The more than fourfold increase in iatrogenic POI (7.6% to 34.2%) is attributed to improved oncologic treatments and rising numbers of gynecologic surgeries [1]. Simultaneously, the doubling of autoimmune-associated POI (8.7% to 18.9%) likely reflects enhanced detection methods and understanding of autoimmune mechanisms, while genetic causes have remained stable [1].
Genetic Architecture of POI
Inheritance Patterns and Genetic Mechanisms
POI demonstrates remarkable genetic heterogeneity, with involvement of more than 100 genes spanning various chromosomal regions and biological processes [2]. The genetic architecture extends beyond simple monogenic inheritance to include digenic, oligogenic, and polygenic models [2]. A 2025 study utilizing a 163-gene NGS panel identified causal genetic anomalies in 57.1% (16/28) of idiopathic POI patients, with single nucleotide variations (28.6%) and copy number variations (3.6%) representing major contributors [3].
Table 2: Major Genetic Causes and Mechanisms in POI
| Genetic Category | Key Genes/Examples | Prevalence & Notes | Associated Phenotypes |
|---|---|---|---|
| Chromosomal Abnormalities | Turner Syndrome (45,X and variants), X-structural anomalies | ~12-13% of POI cases; more frequent in primary amenorrhea (21.4%) [1] | Often syndromic with extra-ovarian features |
| FMR1 Premutations | FMR1 (55-200 CGG repeats) | 20-30% of carriers develop FXPOI; highest risk with 70-100 repeats [1] | Isolated ovarian insufficiency |
| Meiosis & DNA Repair Genes | MSH4, MSH5, HFM1, SPIDR, SMC1B, STAG3 | Account for ~14.4% of cases in large cohort studies [4] | Mostly isolated POI |
| Transcription Factors | NOBOX, FOXL2, FIGLA, SOHLH1, NR5A1 | FOXL2 variants found in 3.2% of cases [4] | Both isolated and syndromic forms |
| Folliculogenesis Genes | BMP15, GDF9, FSHR, BMPR2 | Isolated ovarian insufficiency |
Key Pathogenic Genes and Pathways
The functional classification of POI-associated genes reveals critical biological pathways essential for ovarian function:
- Meiosis and DNA Repair: Genes including MSH4, MSH5, HFM1, SPIDR, SMC1B, and STAG3 are crucial for homologous recombination and meiotic progression [4] [2]. A 2023 study of 500 POI patients identified pathogenic variants in these genes in 14.4% of cases, with digenic inheritance observed in MSH4 and MSH5 [4].
- Transcription Regulation: NOBOX and FOXL2 represent significant transcription factors, with FOXL2 variants reaching 3.2% prevalence in large cohorts [4]. Notably, specific FOXL2 variants (p.R349G) can cause isolated ovarian insufficiency without the characteristic blepharophimosis-ptosis-epicanthus inversus syndrome [4].
- Follicular Development and Function: BMP15, GDF9, FIGLA, and FSHR regulate folliculogenesis, with mutations leading to accelerated follicular atresia [2].
Diagram Title: Genetic Pathways in POI Pathogenesis
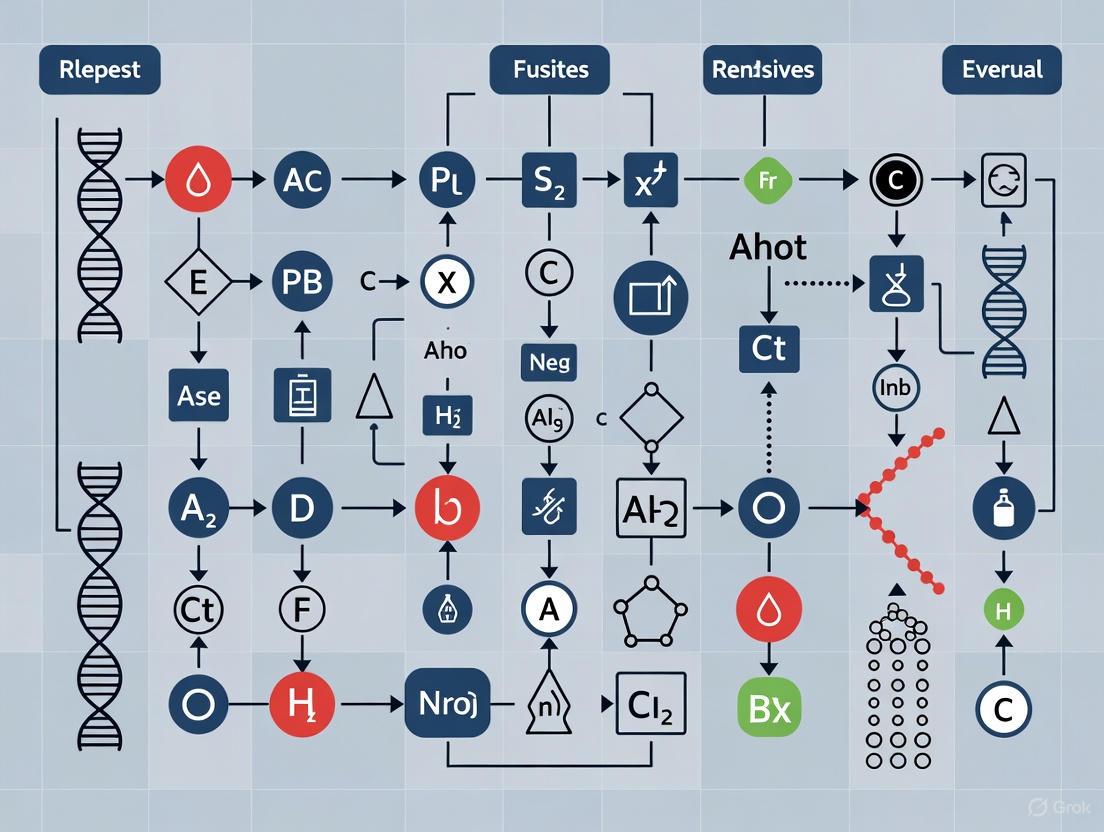
Comprehensive NGS Panel Methodology for POI
Patient Selection and Diagnostic Criteria
The application of a 163-gene NGS panel requires careful patient selection and standardized diagnostic criteria. The protocol should include women presenting with:
- Primary amenorrhea (absence of menarche by age 15) or secondary amenorrhea (cessation of menses for ≥4 months)
- Age of onset <40 years
- Elevated FSH levels >25 IU/L on two occasions at least 4 weeks apart
- Exclusion of other etiologies (karyotype abnormalities, FMR1 premutations, autoimmune, or iatrogenic causes) [3]
Recent studies demonstrate that patients with primary amenorrhea (14.3% in cohort studies) and those with positive family history (39.3%) show higher diagnostic yields [3].
Laboratory Workflow and Technical Specifications
The NGS panel implementation follows a rigorous technical workflow with multiple quality control checkpoints:
Diagram Title: NGS Panel Analysis Workflow
Key technical specifications include:
- DNA Extraction: QIAsymphony DNA midi kits on QIAsymphony system (Qiagen) [3]
- CNV Detection: Oligonucleotide array-CGH using SurePrint G3 Human CGH Microarray 4×180K technology (Agilent Technologies) with 60kb minimum resolution [3]
- Library Preparation: SureSelect XT-HS reagents (Agilent Technologies) with custom capture design of 163 POI-associated genes [3]
- Sequencing Platform: NextSeq 550 system (Illumina) [3]
- Bioinformatics Analysis: Alissa Align&Call v1.1 and Alissa Interpret v5.3 software (Agilent Technologies) [3]
Variant Interpretation and Classification
Variant interpretation follows established American College of Medical Genetics (ACMG/AMP) guidelines with specific refinements:
- Population frequency filtering: Variants with frequency >0.1% in gnomAD and 1000 Genomes Project are excluded [3] [4]
- In silico prediction tools: MetaSVM, CADD, and DANN scores for functional impact prediction [4]
- Variant classification: Five-tier system (Benign, Likely Benign, Variant of Uncertain Significance - VUS, Likely Pathogenic, Pathogenic) [3]
- Segregation analysis: Pedigree haplotype analysis for confirming compound heterozygous variants [4]
Research Reagent Solutions for POI Genetic Studies
Table 3: Essential Research Reagents and Platforms for POI Genetic Studies
| Category | Specific Product/Platform | Function/Application | Manufacturer |
|---|---|---|---|
| DNA Extraction | QIAsymphony DNA Midi Kits | Automated nucleic acid purification | Qiagen |
| Array CGH | SurePrint G3 Human CGH Microarray 4×180K | Genome-wide CNV detection | Agilent Technologies |
| CGH Software | CytoGenomics v5.0 | CNV data analysis and visualization | Agilent Technologies |
| NGS Target Capture | SureSelect XT-HS Custom Design | Sequence enrichment of POI gene panels | Agilent Technologies |
| NGS Sequencing | NextSeq 550 System | High-throughput sequencing | Illumina |
| Variant Analysis | Alissa Align&Call, Alissa Interpret | Variant calling and interpretation | Agilent Technologies |
| CNV Analysis | Cartagenia Bench Lab CNV v5.1 | Clinical CNV interpretation | Agilent Technologies |
| Variant Classification | ACMG/AMP Guidelines | Standardized pathogenicity assessment | ClinGen |
Clinical Correlations and Genotype-Phenotype Associations
Genetic findings demonstrate distinct correlations with clinical presentation. Patients with oligogenic variants (digenic or multigenic) often present with more severe phenotypes, including delayed menarche, early POI onset, and higher prevalence of primary amenorrhea compared to those with monogenic variants [4]. A 2023 study of 500 POI patients revealed that 1.8% (9/500) with digenic/multigenic variants exhibited this severe clinical profile [4].
Interestingly, some genotype-phenotype correlations challenge traditional assumptions. For instance, specific FOXL2 variants can cause isolated ovarian insufficiency without the characteristic syndromic features [4]. Furthermore, the same genetic variant may manifest with different clinical severity within families, supporting the role of modifier genes and oligogenic inheritance [2].
The implementation of comprehensive NGS panels representing 163 POI-associated genes has dramatically improved our understanding of POI heterogeneity, reducing idiopathic cases from >70% to approximately 37% [1] [3]. The 57.1% diagnostic yield achieved through combined array-CGH and NGS approaches demonstrates the power of comprehensive genetic assessment in elucidating POI pathogenesis [3].
Future directions should focus on:
- Functional validation of VUS classifications through experimental assays
- Expansion of gene panels to include newly discovered candidates
- Integration of oligogenic scoring systems to account for complex inheritance
- Long-term correlation of genetic subtypes with therapeutic responses and health outcomes
The established protocol provides a robust framework for genetic diagnosis of POI, enabling personalized risk assessment, familial screening, and targeted management of this complex condition.
Premature Ovarian Insufficiency (POI) is a clinically heterogeneous disease characterized by the loss of ovarian function before the age of 40, affecting approximately 1-3.7% of women and representing a significant cause of female infertility [3] [5]. The condition manifests through primary or secondary amenorrhea, elevated gonadotropin levels, and estrogen deficiency, often leading to serious long-term health complications including osteoporosis and cardiovascular disease [3] [6]. While POI etiologies encompass iatrogenic, autoimmune, and genetic causes, nearly 70% of cases remain idiopathic, underscoring the critical need for advanced molecular diagnostics to elucidate underlying pathophysiological mechanisms [3].
Next-generation sequencing (NGS) technologies have revolutionized the genetic diagnosis of POI, enabling comprehensive analysis of multiple candidate genes simultaneously. The 163-gene panel represents a targeted approach focusing on genes with known or suspected associations with ovarian function, providing a cost-effective alternative to broader whole-exome or whole-genome sequencing while delivering manageable datasets for clinical interpretation [3] [7]. This panel specifically interrogates three fundamental biological pathways essential for ovarian development and function: meiosis, DNA repair, and folliculogenesis.
The integration of this targeted genetic screening into clinical practice enables unprecedented personalization of patient management, from fertility counseling to the prevention and treatment of associated comorbidities [5]. This application note delineates the key pathways, experimental protocols, and analytical frameworks underpinning the 163-gene panel, providing researchers and clinicians with a comprehensive resource for implementing this powerful diagnostic tool.
Key Biological Pathways in POI
Meiosis and DNA Repair Pathways
Meiosis, the specialized cell division that generates haploid gametes, represents a cornerstone of female reproductive function, with its proper execution being absolutely essential for the production of viable oocytes. This process is particularly crucial in females, as the entire pool of oocytes is established during fetal development and must remain functionally intact throughout reproductive life [6].
DNA Double-Strand Break (DSB) Formation and Repair: The initiation of meiotic recombination relies on programmed DNA double-strand breaks (DSBs), which are catalyzed by the SPO11 protein in concert with topoisomerase VIBL (TopoVIBL) and additional factors including PRDM9, MEI1, MEI4, REC114, and ANKRD31 [6]. These DSBs are subsequently repaired through two primary mechanisms:
- Homologous Recombination (HR): This high-fidelity repair pathway involves end resection creating 3' single-stranded DNA overhangs, followed by RPA binding and subsequent replacement by RAD51 and its meiotic-specific paralog DMC1. The RAD51/DMC1-nucleoprotein filaments then facilitate strand invasion using homologous templates, with key regulatory components including BRCA2, MCM8-MCM9 helicase complex, MSH4-MSH5 heterodimer, HFM1, and RECQL4 [6].
- Non-Homologous End Joining (NHEJ): This error-prone repair mechanism functions throughout the cell cycle but is particularly active during G1 phase, utilizing a suite of polymerases, nucleases, and ligases to directly rejoin broken DNA ends [6].
Table 1: Key Meiosis and DNA Repair Genes in the 163-Gene Panel and Their Associated Functions
| Gene | Pathway | Biological Function | POI Association Evidence |
|---|---|---|---|
| SPO11 | Meiosis/DSB Formation | Catalytic subunit for programmed DNA double-strand break formation | Established in multiple studies [6] |
| DMC1 | Meiosis/Homologous Recombination | Meiotic-specific recombinase facilitating strand invasion | Pathogenic variants identified in POI patients [3] [6] |
| RAD51 | Homologous Recombination | Facilitates DNA strand exchange in mitotic and meiotic cells | Supported by functional studies [6] |
| MSH4 | Meiotic Recombination | Forms heterodimer with MSH5 to stabilize Holliday junctions | Mutations reported in POI cohorts [6] [5] |
| MCM8 | DNA Repair/HR | Helicase component involved in DNA replication and repair | Strong association with POI pathogenesis [6] |
| MCM9 | DNA Repair/HR | Partners with MCM8 in DNA repair machinery | Biallelic mutations cause POI [6] |
| BRCA2 | DNA Repair/HR | Mediates RAD51 loading onto single-stranded DNA | Tumor susceptibility genes with POI phenotype [5] |
| HELQ | DNA Repair | Helicase Q involved in DNA interstrand cross-link repair | Newly identified in POI pathogenesis [5] |
| SWI5 | DNA Repair/HR | Facilitates RAD51 focus formation in meiosis | Newly identified in POI pathogenesis [5] |
The critical importance of DNA repair mechanisms in ovarian function is evidenced by the observation that mutations in key DSB repair genes can trigger accelerated follicular atresia or oocyte apoptosis, ultimately depleting the ovarian reserve and culminating in POI [6]. Recent research has identified several new DNA repair genes in POI pathogenesis, including C17orf53 (HROB), HELQ, and SWI5, which are associated with high chromosomal fragility when mutated [5].
Folliculogenesis and Ovarian Development Pathways
Folliculogenesis encompasses the complex, multi-stage process of ovarian follicle development, from primordial follicle recruitment through to ovulation. This pathway involves precise coordination of oocyte growth and maturation alongside the proliferation and differentiation of surrounding granulosa and theca cells [3] [5].
Key Signaling Pathways and Molecular Regulators:
- TGF-β Superfamily Signaling: This pivotal pathway includes bone morphogenetic proteins (BMPs) such as BMP15, GDF9, and anti-Müllerian hormone (AMH), which regulate follicular development and ovulation quota. Mutations in BMP15 and GDF9 have been established as causative factors in POI, disrupting normal follicular maturation [3].
- Transcription Factors: Genes including FIGLA and NOBOX function as master regulators of oocyte-specific gene expression programs. Biallelic mutations in FIGLA have been identified as pathogenic in patients with primary amenorrhea, highlighting their non-redundant roles in ovarian development [3].
- Receptor Signaling: Estrogen receptors (ESR2), bone morphogenetic protein receptors (BMPR1A, BMPR1B, BMPR2), and other signaling components transduce extracellular signals to coordinate follicular growth and maturation [5].
- Novel Pathways: Recent evidence has implicated previously unrecognized pathways in POI pathogenesis, including NF-κB signaling, post-translational regulation, and mitophagy (mitochondrial autophagy), offering new therapeutic targets for intervention [5].
Table 2: Key Folliculogenesis Genes in the 163-Gene Panel and Their Functional Roles
| Gene | Molecular Function | Role in Ovarian Function | Clinical Manifestation When Mutated |
|---|---|---|---|
| FIGLA | Transcription Factor | Regulates oocyte-specific gene expression | Primary amenorrhea (homozygous mutations) [3] |
| BMP15 | Growth Factor | Oocyte-derived factor promoting follicular development | Isolated or syndromic POI [3] |
| GDF9 | Growth Factor | Modulates granulosa cell proliferation and differentiation | POI with variable expressivity [3] |
| NOBOX | Transcription Factor | Essential for primordial follicle activation | POI with progressive follicular depletion [3] |
| ESR2 | Hormone Receptor | Mediates estrogen signaling in ovarian tissue | Impaired follicular growth and ovulation [5] |
| BMPR1B | Receptor Signaling | Transduces BMP signaling in granulosa cells | Disrupted folliculogenesis [5] |
| ATG7 | Autophagy | Regulates mitophagy and cellular quality control | Premature follicular depletion [5] |
| NLRP11 | Inflammation/NF-κB | Regulates inflammatory responses in ovarian tissue | Newly associated with POI [5] |
Quantitative Detection Rates and Clinical Yield
The diagnostic yield of the 163-gene panel has been systematically evaluated in clinical cohorts, providing evidence-based metrics for its implementation. A recent study of 28 idiopathic POI patients (14.3% with primary amenorrhea, 85.7% with secondary amenorrhea) demonstrated a 57.1% overall detection rate of pathogenic genetic anomalies, comprising copy number variations (CNVs), single nucleotide variations (SNVs), and indel mutations [3].
Table 3: Diagnostic Yield of the 163-Gene Panel in POI Patients
| Genetic Finding | Detection Rate | Number of Patients | Clinical Implications |
|---|---|---|---|
| Causal CNV | 3.6% (1/28) | 1 | Often associated with syndromic features; requires comprehensive phenotyping |
| Causal SNV/Indel | 28.6% (8/28) | 8 | Enables precise genetic counseling and familial screening |
| Variants of Uncertain Significance (VUS) | 25% (7/28) | 7 | May be reclassified with additional evidence; cautious interpretation required |
| Any Genetic Anomaly | 57.1% (16/28) | 16 | Facilitates personalized management of comorbidities and fertility planning |
| Familial History Positive | 39.3% (11/28) | 11 | Supports autosomal dominant or X-linked inheritance patterns |
Notably, the study identified a higher prevalence of familial POI cases (39.3%) than previously recognized in the literature (historically estimated at 12-31%), suggesting a stronger genetic component than conventionally appreciated [3]. Furthermore, comprehensive genetic analyses have revealed that in 8.5% of cases, POI represents the sole presenting symptom of a multi-system genetic disorder, emphasizing the importance of genetic diagnosis for comprehensive patient management [5].
The integration of multi-omics approaches is further enhancing our understanding of POI pathogenesis. Recent investigations have identified STAT3 as a hub gene in hypertrophic cardiomyopathy pathways [8], illustrating how cross-disciplinary analyses can reveal novel molecular insights. Additionally, bioinformatics approaches such as Weighted Gene Correlation Network Analysis (WGCNA) have proven valuable for identifying key pathways and circulating markers in other complex diseases [9], suggesting their potential application to POI research.
Experimental Protocol for 163-Gene Panel Analysis
Sample Preparation and Quality Control
Materials and Equipment:
- QIAsymphony DNA Mid Kits (Qiagen)
- QIAsymphony automated nucleic acid extraction system
- Agarose gel electrophoresis system
- Spectrophotometer (NanoDrop or equivalent)
- Qubit fluorometer with dsDNA HS Assay Kit
Procedure:
- DNA Extraction: Isolate genomic DNA from peripheral blood samples using QIAsymphony DNA Mid Kits according to manufacturer's protocols [3].
- DNA Quantification: Measure DNA concentration using both spectrophotometric (A260/A280 ratio of 1.8-2.0) and fluorometric methods to ensure accurate quantification.
- Quality Assessment: Verify DNA integrity by agarose gel electrophoresis, ensuring high-molecular-weight DNA without significant degradation.
- Normalization: Dilute samples to working concentration of 10-20 ng/μL in nuclease-free water or TE buffer.
Targeted Enrichment and Library Preparation
Materials and Equipment:
- Custom SureSelect XT-HS Target Enrichment System (Agilent Technologies)
- Magnis NGS Prep System (Agilent Technologies)
- Thermal cycler with heated lid
- Magnetic separation stand
- Size selection beads (AMPure XP or equivalent)
Procedure:
- Library Preparation: Fragment 100-200 ng of genomic DNA and add Illumina-compatible adapters using SureSelect XT-HS reagents according to manufacturer's specifications [3].
- Target Enrichment: Hybridize library DNA with custom biotinylated RNA baits (163-gene panel) for 16-24 hours at 65°C with agitation [3].
- Magnetic Capture: Bind bait-library hybrids to streptavidin-coated magnetic beads, followed by stringent washing to remove non-specifically bound DNA.
- PCR Amplification: Amplify captured libraries using index primers to enable sample multiplexing (10-12 cycles typically sufficient).
- Library Quantification: Assess library concentration by qPCR and fragment size distribution by bioanalyzer or tape station.
Next-Generation Sequencing
Materials and Equipment:
- NextSeq 550 sequencing system (Illumina)
- NextSeq 500/550 High Output Kit v2.5 (150 cycles)
- PhiX Control v3 adapter-ligated library
Procedure:
- Pool Normalization: Combine indexed libraries in equimolar ratios, typically aiming for 100-200 million clusters per run.
- Quality Control: Include 1% PhiX control library to monitor sequencing performance and assess base calling accuracy.
- Cluster Generation: Load pooled libraries onto the flow cell and perform bridge amplification according to Illumina's standard protocols.
- Sequencing: Run paired-end 2×75 bp or 2×150 bp sequencing to achieve minimum 100× average coverage across all targets.
- Data Output: Demultiplex samples and generate FASTQ files for downstream analysis.
Bioinformatic Analysis
Computational Tools:
- Alissa Align&Call v1.1 and Alissa Interpret v5.3 (Agilent Technologies)
- Feature Extraction and CytoGenomics software v5.0 (Agilent Technologies)
- Cartagenia Bench Lab CNV software v5.1 (Agilent Technologies)
- gnomAD, DECIPHER, ClinVar, HGMD databases
Procedure:
- Sequence Alignment: Map sequencing reads to reference genome (GRCh37/hg19) using Burrows-Wheeler Aligner (BWA) or similar aligner.
- Variant Calling: Identify single nucleotide variants (SNVs) and small insertions/deletions (indels) using GATK HaplotypeCaller or similar algorithm.
- Variant Annotation: Annotate variants with population frequency, in silico prediction scores, and clinical interpretations from curated databases.
- Copy Number Variation Analysis: Detect exon-level CNVs using read depth-based algorithms with comparison to reference samples.
- Variant Filtering and Prioritization:
- Remove common polymorphisms (gnomAD AF > 0.01)
- Prioritize protein-altering variants (missense, nonsense, splice-site, indels)
- Apply ACMG/AMP guidelines for variant classification [3]
- Validation: Confirm pathogenic and likely pathogenic variants by Sanger sequencing.
Pathway Visualization and Analytical Framework
Diagram 1: Key biological pathways implicated in the 163-gene panel for POI, highlighting the interconnections between meiosis, DNA repair, and folliculogenesis processes.
Research Reagent Solutions
Table 4: Essential Research Reagents and Platforms for 163-Gene Panel Implementation
| Reagent/Platform | Vendor | Application | Key Features |
|---|---|---|---|
| SureSelect XT-HS Custom | Agilent Technologies | Target Enrichment | Custom capture design for 163 genes; optimized for FFPE, blood, and saliva samples |
| Illumina DNA Prep with Enrichment | Illumina | Library Preparation | Rapid, flexible targeted sequencing library prep for genomic DNA |
| NextSeq 550 System | Illumina | Sequencing | Mid-throughput sequencing with fast turnaround time |
| QIAsymphony DNA Mid Kits | Qiagen | Nucleic Acid Extraction | Automated, high-quality DNA extraction from peripheral blood |
| Alissa Interpret | Agilent Technologies | Variant Interpretation | ACMG-compliant variant classification and reporting |
| Cartagenia Bench Lab CNV | Agilent Technologies | CNV Analysis | Sensitive detection of copy number variations from NGS data |
| DesignStudio Software | Illumina | Panel Design | Online tool for optimizing custom targeted enrichment designs |
| AmpliSeq for Illumina | Illumina | Amplicon Sequencing | PCR-based targeting ideal for smaller gene panels (<50 genes) |
The 163-gene panel represents a significant advancement in the molecular diagnosis of Premature Ovarian Insufficiency, enabling comprehensive assessment of key biological pathways including meiosis, DNA repair, and folliculogenesis. With a diagnostic yield exceeding 57% in idiopathic cases, this targeted approach provides valuable insights for patient management, familial counseling, and therapeutic decision-making [3]. The integration of multi-omics strategies continues to expand our understanding of POI pathogenesis, revealing novel genes and pathways while highlighting the potential for personalized treatment approaches tailored to an individual's genetic profile [5] [10].
As research progresses, the refinement of gene panels and analytical frameworks will further enhance our ability to diagnose and manage this complex disorder, ultimately improving outcomes for affected women through precision medicine approaches.
Premature ovarian insufficiency (POI) is a clinically heterogeneous disorder characterized by the loss of ovarian activity before the age of 40 years, presenting as primary or secondary amenorrhea with elevated follicle-stimulating hormone (FSH) levels greater than 25 IU/L [3] [11]. This condition affects approximately 1% of women under 40, with incidence varying from 1:10,000 women by age 20 to 1:100 women under 40 [3] [11] [12]. POI leads to significant health consequences including infertility, increased risk of osteoporosis, cardiovascular disease, and other conditions associated with estrogen deficiency [3].
While chromosomal abnormalities and FMR1 premutations have long been recognized as genetic causes of POI, recent evidence confirms that autosomal genes play an equally critical role in its pathogenesis [11]. The identification of 57.1% of patients carrying causal genetic variations in a recent study highlights the substantial contribution of autosomal genetic factors to POI etiology [3]. This application note details the implementation and utility of a comprehensive next-generation sequencing (NGS) panel targeting 163 POI-associated genes, providing researchers with validated protocols for identifying autosomal genetic determinants in POI populations.
Table 1: Key Epidemiological and Genetic Features of POI
| Parameter | Value/Range | Clinical Significance |
|---|---|---|
| Prevalence <40 years | 1% | Significant impact on reproductive health and quality of life [3] |
| Genetic Etiology | 20-25% | Substantial portion with identifiable genetic causes [11] |
| Idiopathic Cases | ~70% | Majority without known etiology, potential for new gene discovery [3] |
| Familial Aggregation | 12-31% | Strong heritable component [3] [11] |
| Successful NGS Detection | 57.1% | High diagnostic yield with comprehensive genetic testing [3] |
Autosomal Genetic Landscape in POI
Biological Pathways and Mechanisms
Autosomal genes implicated in POI pathogenesis participate in diverse biological processes essential for normal ovarian function, including:
- Meiosis and DNA Repair: Genes such as MCM8, DMC1, and STAG3 are crucial for chromosomal stability, homologous recombination, and DNA break repair during oocyte development [3] [11].
- Folliculogenesis and Ovulation: Transcription factors including NOBOX, FIGLA, and FOXL2 regulate primordial follicle activation, granulosa cell differentiation, and follicular development [3] [11] [12].
- Steroidogenesis and Hormone Signaling: Genes such as CYP17A1, CYP19A1, and FSHR encode proteins critical for hormone biosynthesis and response [12].
- Metabolic Processes: Variants in genes like PMM2 can disrupt cellular functions indirectly affecting ovarian reserve [3].
The inheritance patterns for these autosomal genes include both dominant and recessive modes, with some genes (e.g., FSHR, LMNA) associated with either pattern depending on the specific variant [13] [12]. This pathogenic diversity underscores the necessity for comprehensive genetic analysis in POI patients.
Key Autosomal Genes in POI Pathogenesis
Table 2: Selected Autosomal Genes and Their Roles in POI Pathogenesis
| Gene | Inheritance Pattern | Primary Ovarian Function | Reported Variant Types |
|---|---|---|---|
| FIGLA | Autosomal Recessive | Oocyte development and primordial follicle activation | Frameshift, nonsense [3] |
| NOBOX | AD/AR | Early folliculogenesis, oocyte-specific transcription | Missense, loss-of-function [11] [12] |
| FOXL2 | Autosomal Dominant | Granulosa cell differentiation, ovary maintenance | Nonsense, frameshift, missense [12] |
| STAG3 | Autosomal Recessive | Meiotic cohesin component, chromosome segregation | Loss-of-function [12] |
| BMP15 | X-linked | Oocyte factor for follicular development | Missense, regulatory [12] |
| NR5A1 | Autosomal Dominant | Steroidogenic factor, adrenal and gonadal development | Haploinsufficiency, missense [12] |
Comprehensive NGS Panel Design and Workflow
Panel Configuration and Target Regions
The 163-gene NGS panel employs a custom capture design encompassing genes with established or suspected roles in ovarian function [3]. The panel design includes:
- Complete coding regions of all target genes with flanking intronic sequences to capture splice-site variants
- Non-coding pathogenic variants located up to ±20 base pairs from exon-intron boundaries
- Copy Number Variation (CNV) detection capability through comparative read depth analysis
- Unique Molecular Indexes (UMIs) to reduce PCR amplification artifacts and improve detection accuracy [14]
This comprehensive approach enables simultaneous detection of single nucleotide variants (SNVs), small insertions/deletions (indels), and larger copy number variations, providing a complete genetic profile from a single assay [3].
Experimental Protocol: Library Preparation and Sequencing
Table 3: Key Research Reagent Solutions for NGS Panel Implementation
| Reagent/Equipment | Function | Specifications/Alternatives |
|---|---|---|
| SureSelect XT HS Kit (Agilent) | Library preparation | Optimized for FFPE DNA, low input capability [3] [14] |
| QIAsymphony DNA Mid Kits (Qiagen) | DNA extraction | High-quality DNA from blood/saliva [3] |
| NextSeq 550 System (Illumina) | Sequencing platform | 2 × 75 bp paired-end reads recommended [3] |
| Magnis System (Agilent) | Library preparation | Automated system for processing [3] |
| Covaris ME220 | DNA shearing | Ultrasonicator for controlled fragmentation [14] |
| Agencourt AMPure XP Beads (Beckman) | Size selection | PCR purification and clean-up [14] |
Sample Requirements and Quality Control
- DNA Source: Peripheral blood, saliva, or extracted DNA
- Minimum Input: 50-200 ng genomic DNA
- Quality Metrics: DNA Integrity Number (DIN) >2.0, though >3.0 recommended [14]
- Quantification: Fluorometric methods (e.g., Qubit Fluorometer) for accurate DNA concentration measurement [14]
Library Preparation Protocol
- DNA Fragmentation: Fragment 50-200 ng genomic DNA to ~200 bp using focused-ultrasonication [14].
- End Repair and A-Tailing: Repair fragment ends and add adenosine overhangs using SureSelect XT HS reagents.
- Adapter Ligation: Ligate adapters incorporating unique molecular indexes (UMIs) for duplicate removal and error suppression.
- PCR Amplification: Amplify libraries with 10-16 cycles using adapter-specific primers.
- Target Enrichment: Hybridize with biotinylated capture probes specific to the 163-gene panel.
- Post-Capture Amplification: Enrich captured libraries with 12 PCR cycles followed by bead-based purification [14].
Sequencing and Data Analysis
- Sequencing Configuration: 2 × 75 bp paired-end reads on Illumina NextSeq 550 or similar platform
- Minimum Coverage: 100x recommended, with >95% of target bases ≥20x coverage [3]
- Bioinformatics Pipeline: Alignment to reference genome (GRCh37/38), variant calling, and annotation using tools such as Alissa Align&Call and Alissa Interpret [3]
Figure 1: NGS Analysis Workflow for POI Genetic Testing. The process from sample collection to final variant reporting includes multiple quality control checkpoints to ensure data reliability.
Analytical Validation and Performance Metrics
Sensitivity and Specificity Measurements
Rigorous validation of the 163-gene panel demonstrates robust performance characteristics comparable to established NGS panels [3] [15]. Based on orthogonal validation studies:
- Sensitivity: 98.23% for variant detection at 95% confidence interval [15]
- Specificity: 99.99% for distinguishing true variants from background noise [15]
- Limit of Detection: Reliable detection of variants at ≥2.9% variant allele frequency (VAF) [15]
- Reproducibility: 99.99% consistency between replicate algorithm runs [15]
Variant Classification Framework
Identified variants are classified according to American College of Medical Genetics (ACMG) guidelines:
- Class 1 (Benign): No association with POI pathogenesis
- Class 2 (Likely Benign): Low probability of pathogenicity
- Class 3 (VUS): Variant of Uncertain Significance requiring further investigation
- Class 4 (Likely Pathogenic): High probability of pathogenicity
- Class 5 (Pathogenic): Established disease-causing variants [3]
Table 4: Performance Metrics of the 163-Gene POI NGS Panel
| Performance Parameter | Result | Method of Assessment |
|---|---|---|
| Analytical Sensitivity | 98.23% (95% CI) | Comparison with orthogonal methods [15] |
| Analytical Specificity | 99.99% (95% CI) | False positive rate evaluation [15] |
| Reproducibility | 99.99% | Inter-run precision [15] |
| Repeatability | 99.99% | Intra-run precision [15] |
| Minimum VAF | 2.9% | Limit of detection analysis [15] |
| Diagnostic Yield | 57.1% | Clinical validation in 28 POI patients [3] |
Research Applications and Clinical Translation
Integration with Other Genomic Analyses
For comprehensive POI genetic assessment, the autosomal gene NGS panel should be complemented with:
- Karyotype Analysis: Detection of X chromosomal abnormalities and mosaicism [11]
- FMR1 CGG Repeat Analysis: Identification of premutation alleles (55-200 repeats) associated with fragile X-associated POI [11]
- Array CGH: Genome-wide detection of copy number variations beyond the target genes [3]
The combined diagnostic approach significantly improves the overall detection rate of pathogenic variations, with one study reporting causal CNVs in 3.6% of patients and causal SNVs/indels in 28.6% [3].
Implications for Drug Development and Therapeutic Strategies
Identification of autosomal gene defects in POI creates opportunities for targeted therapeutic interventions:
- Pathway-Specific Therapeutics: Genes involved in follicular development (e.g., BMP15, GDF9) represent potential targets for ovarian stimulation protocols [11]
- DNA Repair Modulation: Patients with mutations in DNA repair genes (e.g., MCM8, DMC1) may benefit from treatments that reduce oxidative stress or enhance DNA repair mechanisms [3]
- Hormone Signaling Targets: Variants in steroidogenesis genes (e.g., CYP17A1, CYP19A1) inform personalized hormone replacement strategies [12]
Figure 2: From Genetic Findings to Therapeutic Strategies. Autosomal gene variants are categorized by pathogenic mechanism, enabling development of targeted interventions based on affected biological pathways.
The implementation of a comprehensive NGS panel targeting 163 POI-associated autosomal genes represents a significant advancement in reproductive genetics research. The validated protocols and analytical frameworks presented in this application note provide researchers with robust tools for elucidating the substantial contribution of autosomal genes to POI pathogenesis. With a diagnostic yield exceeding 57% in idiopathic cases, this approach substantially reduces the number of cases classified as unexplained [3]. The integration of these genetic findings into both clinical management and drug development pipelines promises to advance personalized medicine approaches for women with POI, ultimately improving reproductive outcomes and long-term health for affected individuals.
Premature ovarian insufficiency (POI) is a clinically heterogeneous disorder characterized by the loss of ovarian function before the age of 40, affecting approximately 1–3.7% of women [16] [17] [18]. It is defined by oligomenorrhea or amenorrhea for at least 4 months, with elevated follicle-stimulating hormone (FSH) levels (>25 IU/L) and low estradiol [17]. The etiological landscape of POI encompasses chromosomal abnormalities, autoimmune disorders, iatrogenic causes, and genetic defects, though up to 70% of cases remain idiopathic [3] [16]. A significant heritable component is evidenced by familial clustering observed in 12–31% of cases [3], with molecular causes identified in 20–25% of patients [3] [19]. Next-generation sequencing (NGS) technologies have revolutionized the identification of genetic defects underlying POI, enabling molecular diagnosis in a substantial proportion of previously idiopathic cases. This application note details the implementation and validation of an NGS panel targeting 163 POI-associated genes, providing researchers with a comprehensive framework for genetic investigation of this complex disorder.
Genetic Architecture of POI
Diagnostic Yield of Genetic Testing
Table 1: Diagnostic Yield of Genetic Investigations in POI
| Investigation Method | Cohort Size | Diagnostic Yield | Key Findings | Citation |
|---|---|---|---|---|
| Array-CGH + NGS (163 genes) | 28 patients | 57.1% (16/28) | 1 causal CNV; 8 causal SNVs/indels (28.6%); 7 VUS | [3] |
| Targeted NGS (28 genes) | 500 patients | 14.4% (72/500) | 61 P/LP variants in 19 genes; 58 novel variants | [19] |
| Targeted NGS (295 genes) | 64 patients | 75% (48/64) | Oligogenic involvement: 17% with 2 variants, 14% with 3 variants | [20] |
| Whole Genome Sequencing (FXPOI) | 114 PM carriers | 8% variance explained | PRS based on natural menopause variants | [21] |
Spectrum of Genetic Variants
The genetic architecture of POI encompasses monogenic, oligogenic, and polygenic contributions. Chromosomal abnormalities, particularly X-chromosome anomalies and FMR1 premutations, represent the most frequently identified genetic causes [16]. NGS studies have identified pathogenic variants in numerous genes involved in key biological processes:
- Meiosis and DNA Repair: NBN, MSH4, MSH5, HFM1, SPIDR [22] [16] [19]
- Folliculogenesis: NOBOX, FIGLA, BMP15, GDF9 [16] [19]
- Transcription Factors: FOXL2, NR5A1, SOHLH1 [19]
- Hormone Signaling: FSHR, BMPR2, AMH [19]
Notably, recent evidence supports an oligogenic inheritance model in which the cumulative effect of variants in multiple genes contributes to disease severity and presentation [20]. Patients with digenic or multigenic variants often present with more severe phenotypes, including delayed menarche, earlier POI onset, and higher prevalence of primary amenorrhea [19].
Experimental Protocols
Targeted NGS Panel for POI
Panel Design and Target Selection
- Gene Selection Criteria: Curate 163 genes with established or putative roles in ovarian development and function, including genes involved in gonadal development, meiosis, folliculogenesis, and steroidogenesis [3].
- Target Enrichment: Design a custom capture using SureSelect XT-HS reagents (Agilent Technologies) targeting all coding exons and flanking splice sites (±50 bp) [3].
- Quality Control: Verify panel specificity and sensitivity using reference samples with known variants in POI-associated genes.
Library Preparation and Sequencing
- DNA Extraction: Extract genomic DNA from peripheral blood using QIAsymphony DNA midi kits (Qiagen) [3].
- Library Preparation:
- Fragment 50-100 ng of genomic DNA
- Perform end-repair, A-tailing, and adapter ligation
- Amplify library with index primers for sample multiplexing
- Target Capture:
- Hybridize library to biotinylated probes
- Capture target regions using streptavidin-coated magnetic beads
- Wash to remove non-specifically bound DNA
- Sequencing: Sequence on Illumina NextSeq 550 system (2×150 bp paired-end reads) to achieve minimum 50× coverage for >90% of target regions [3].
Data Analysis and Variant Interpretation
- Primary Analysis:
- Demultiplex raw sequencing data
- Align reads to reference genome (GRCh37/hg19) using Alissa Align&Call v1.1 (Agilent) [3]
- Variant Calling:
- Call single nucleotide variants (SNVs) and small insertions/deletions (indels)
- Filter variants with quality score <20 and read depth <15× [22]
- Variant Filtering and Annotation:
- Filter against population databases (gnomAD, 1000 Genomes) with frequency threshold <0.1%
- Annotate functional impact using SnpEff
- Predict pathogenicity with multiple algorithms (PolyPhen-2, SIFT, MutationTaster) [22]
- Variant Classification: Classify variants according to ACMG/AMP guidelines [3]
Functional Validation of Identified Variants
Luciferase Reporter Assays
To validate the functional impact of identified variants in transcription factors such as FOXL2:
Plasmid Construction:
- Clone wild-type and mutant cDNA sequences into mammalian expression vectors
- Subclone promoter regions of target genes (e.g., CYP17A1, CYP19A1) into luciferase reporter vectors [19]
Cell Transfection:
- Culture appropriate cell lines (e.g., KGN, COS-7)
- Co-transfect with expression vectors and reporter constructs using lipid-based transfection reagents
- Include Renilla luciferase vector for normalization
Luciferase Assay:
- Harvest cells 48 hours post-transfection
- Measure firefly and Renilla luciferase activities using dual-luciferase reporter assay system
- Calculate relative luciferase activity as ratio of firefly to Renilla luminescence [19]
Pedigree Analysis and Segregation Studies
- Family Recruitment: Recruit available first-degree relatives of probands with candidate pathogenic variants.
- DNA Collection: Extract DNA from peripheral blood of family members.
- Variant Confirmation:
- Validate variants by Sanger sequencing in all available family members
- Perform haplotype analysis for compound heterozygous variants [19]
- Segregation Analysis: Confirm co-segregation of variant(s) with POI phenotype across generations.
Key Biological Pathways in POI Pathogenesis
NGS studies have revealed that POI-associated genes converge on several critical biological pathways essential for ovarian function:
Table 2: Major Pathways Implicated in POI Pathogenesis
| Pathway | Key Genes | Biological Function | Citation |
|---|---|---|---|
| Meiosis & DNA Repair | NBN, MSH4, MSH5, HFM1, SPIDR | Homologous recombination, DNA double-strand break repair, meiotic progression | [22] [16] [19] |
| Folliculogenesis | NOBOX, FIGLA, BMP15, GDF9 | Primordial follicle activation, follicle growth and development | [16] [19] |
| Transcription Regulation | FOXL2, NR5A1, SOHLH1 | Ovarian development, steroidogenic enzyme regulation | [19] |
| Extracellular Matrix Remodeling | GJA4, PGRMC1 | Cell-cell communication, follicle microenvironment maintenance | [20] |
| Cell Metabolism & Signaling | SUM01, KRR1, ESR1 | Post-translational modifications, kinase activity, estrogen signaling | [21] [18] |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for POI Genetic Studies
| Reagent/Category | Specific Product Examples | Application in POI Research | Citation |
|---|---|---|---|
| NGS Library Prep | SureSelect XT-HS (Agilent), Nextera Rapid Capture (Illumina) | Target enrichment for gene panels | [3] [20] |
| Sequencing Platforms | NextSeq 550 (Illumina), Magnis (Agilent) | High-throughput sequencing of POI panels | [3] |
| Bioinformatics Tools | Alissa Align&Call (Agilent), Cpipe, GATK | Variant calling, annotation, and filtering | [22] [3] |
| Variant Databases | gnomAD, 1000 Genomes, ClinVar, HGMD | Population frequency and clinical interpretation | [22] [3] |
| Pathogenicity Prediction | PolyPhen-2, SIFT, CADD, MutationTaster | In silico assessment of variant impact | [22] [19] |
| Functional Validation | Dual-Luciferase Reporter Systems (Promega) | Transcriptional activity assays for variants | [19] |
Discussion and Future Perspectives
The implementation of NGS panels for POI has significantly advanced our understanding of its genetic architecture, moving beyond monogenic causes to recognize oligogenic and polygenic contributions. The 163-gene panel demonstrates a diagnostic yield of approximately 28.6% for causal SNVs/indels [3], with emerging evidence that oligogenic interactions contribute to more severe phenotypes [19] [20].
Critical considerations for clinical translation include:
- Variant Interpretation Challenges: The high prevalence of variants of uncertain significance (VUS) necessitates functional studies and segregation analysis [3].
- Pleiotropic Genes: Variants in genes associated with syndromic conditions (e.g., NBN, EIF2B2) can present as isolated POI, requiring careful counseling and potential screening for subclinical features [22].
- Oligogenic Inheritance: The cumulative effect of multiple variants should be considered, particularly in severe cases [20].
- Ancestry Considerations: Current genetic knowledge is primarily based on European and Asian populations, highlighting the need for diverse population studies [23].
Future directions include the integration of polygenic risk scores (PRS) derived from common variants associated with natural age at menopause [21] [23], which explain approximately 8% of the variance in fragile X-associated POI risk [21]. Additionally, multi-omics approaches incorporating transcriptomic, proteomic, and metabolomic data may further elucidate the complex pathophysiology of POI and identify novel therapeutic targets [18].
The NGS panel approach for POI represents a powerful tool for molecular diagnosis, family counseling, and personalized management of this complex disorder, while continuing to expand our understanding of human ovarian biology.
Implementing the NGS Panel: From Bench to Bioinformatic Analysis
Premature ovarian insufficiency (POI) is a clinically heterogeneous disorder characterized by the loss of ovarian function before the age of 40 years, affecting approximately 1-3.7% of the female population [24] [3]. Its etiology is highly complex, with genetic factors contributing to 20-25% of cases [3] [4]. The need for comprehensive genetic diagnosis has led to the development of targeted next-generation sequencing (NGS) panels that enable the efficient and simultaneous analysis of multiple genes associated with ovarian function. This application note details the technical design and validation of an NGS panel targeting 163 POI-associated genes, a approach that has demonstrated a 57.1% diagnostic yield in identifying pathogenic variations in idiopathic POI cases [3].
Targeted NGS panels represent a practical and cost-effective solution for clinical molecular diagnostics, allowing for deep sequencing of specific genomic regions of interest [25]. In the context of POI, which exhibits significant genetic heterogeneity, such panels facilitate the identification of various mutation types, including single nucleotide variations (SNVs), small insertions and deletions (indels), and copy number variations (CNVs) [3]. The design described herein provides researchers and clinical laboratories with a validated framework for implementing genetic testing for POI, ultimately enabling improved patient management, familial screening, and reproductive counseling.
Technical Design Considerations
Capture Technology Selection and Rationale
The choice of capture methodology is fundamental to the performance of any targeted NGS panel. For the 163-gene POI panel, a hybrid capture-based approach using solution-based, biotinylated oligonucleotide probes was employed [3] [25]. This selection was based on several technical advantages that make it particularly suitable for genetic disorders like POI:
- Comprehensive Coverage: Hybrid capture probes are significantly longer than PCR primers, enabling them to tolerate several mismatches in the probe binding site without interfering with hybridization. This design circumvents issues of allele dropout, which can be problematic in amplification-based assays [25].
- Flexibility in Target Region Design: This approach allows for the targeting of entire genomic regions, including exons, introns, and regulatory elements, which is crucial for capturing variants that may reside in non-coding regions or for detecting structural variants where breakpoints often occur in intronic regions [25].
- Multiplexing Capability: Hybrid capture enables efficient processing of multiple samples in parallel, making it suitable for high-throughput clinical applications and cohort studies [3].
The custom capture design was specifically tailored to target 163 genes known or suspected to be involved in ovarian function, including genes participating in meiotic prophase I, folliculogenesis, DNA replication and repair, and various signaling pathways critical for ovarian development and maintenance [3].
Coverage and Sequencing Depth Specifications
Optimal coverage parameters are critical for achieving high sensitivity and specificity in variant detection. The following specifications were established for the POI panel:
Table 1: Coverage and Sequencing Depth Specifications for the POI NGS Panel
| Parameter | Specification | Rationale |
|---|---|---|
| Minimum Depth of Coverage | >100x | Ensures reliable detection of heterozygous variants [25] |
| Target Mean Coverage | >200x | Provides confidence in variant calling and enables detection of low-level mosaicism [3] |
| Uniformity of Coverage | >95% of targets covered at ≥50x | Minimizes gaps in coverage that could lead to missed variants [25] |
| Target Region Size | Custom 163-gene panel | Balanced approach for comprehensive assessment while maintaining cost-effectiveness [3] |
These parameters ensure sufficient depth to detect various variant types with high confidence, including SNVs and small indels, while maintaining cost-effectiveness for clinical implementation. The high uniformity of coverage is particularly important for avoiding false negatives in regions with lower capture efficiency.
Platform Selection and Sequencing Chemistry
The selection of appropriate sequencing technology directly impacts data quality, throughput, and cost. The POI panel was sequenced on a NextSeq 550 system (Illumina) utilizing sequencing by synthesis (SBS) chemistry [3] [26]. This platform offers several advantages for clinical genetic testing:
- High Accuracy: Illumina's SBS chemistry employs reversible dye-terminators that incorporate a single nucleotide per cycle, resulting in a base-level error rate of <0.1% [26] [27].
- Scalable Throughput: The NextSeq 550 system provides an optimal balance between throughput and flexibility, enabling medium-scale sequencing runs that accommodate batch processing of patient samples [3] [26].
- Proven Clinical Utility: Illumina platforms are widely adopted in clinical diagnostics, with extensive validation data and regulatory clearance for many applications [26] [28].
The platform's ability to generate 75-300 base pair paired-end reads was particularly beneficial for the POI panel, as longer reads improve mapping accuracy, facilitate the detection of structural variants, and enable better coverage across regions with high GC content or repetitive elements [26] [27].
Table 2: Comparison of NGS Platforms for Targeted Gene Panel Sequencing
| Platform | Technology | Read Length | Advantages | Limitations |
|---|---|---|---|---|
| Illumina NextSeq | Sequencing by Synthesis | 75-300 bp (paired-end) | High accuracy, proven clinical utility | Higher capital investment [26] [27] |
| Ion Torrent Semiconductor Sequencing | 200-400 bp | Faster run times, lower initial cost | Homopolymer errors [27] | |
| PacBio SMRT | Single Molecule Real-Time Sequencing | 10,000-25,000 bp (long-read) | Excellent for complex structural variants | Higher error rate, lower throughput [27] |
| Oxford Nanopore | Nanopore Sequencing | 10,000-30,000 bp (long-read) | Ultra-long reads, real-time analysis | Higher error rate (up to 15%) [27] |
Experimental Protocol
Sample Preparation and Quality Control
Proper sample preparation and quality assessment are critical pre-analytical steps that significantly impact downstream sequencing success.
- Sample Requirements: The protocol was validated using DNA extracted from peripheral blood samples collected in EDTA tubes. A minimum of 1-3 μg of high-molecular-weight DNA is required, with optimal concentration of 20-50 ng/μL [3].
- DNA Extraction: DNA is extracted using the QIAsymphony DNA midi kits on a QIAsymphony system (Qiagen) according to manufacturer's specifications [3].
- Quality Control: DNA quality and integrity are assessed using multiple methods:
- Spectrophotometric Analysis: A260/A280 ratio between 1.8-2.0 and A260/A230 ratio >2.0.
- Fluorometric Quantification: Using Qubit dsDNA HS Assay Kit for accurate DNA concentration measurement.
- Fragment Analysis: Genomic DNA should show a majority of fragments >10 kb when analyzed by agarose gel electrophoresis.
For solid tissue samples, microscopic review by a certified pathologist is recommended to ensure sufficient tumor/non-tumor content and to guide macrodissection if needed to enrich target cell populations [25].
Library Preparation Workflow
The library preparation process converts genomic DNA into sequencing-ready libraries compatible with the Illumina platform.
- Fragmentation: 100 ng of input DNA is fragmented using acoustic shearing to a target size distribution of 200-300 bp.
- End Repair and A-Tailing: Fragmented DNA undergoes end-repair to generate blunt ends, followed by A-tailing to facilitate adapter ligation.
- Adapter Ligation: Illumina-specific adapters containing index sequences are ligated to the A-tailed fragments to enable sample multiplexing.
- Library Amplification: Adapter-ligated fragments are amplified using 8-10 cycles of PCR to generate sufficient material for capture.
- Library Quality Control: Amplified libraries are quantified using qPCR and fragment size distribution is verified using a Bioanalyzer or TapeStation system.
The process utilizes SureSelect XT-HS reagents (Agilent Technologies) following the manufacturer's recommendations, with modifications to optimize for the custom POI gene panel [3].
Target Enrichment and Sequencing
The target enrichment process specifically captures the genomic regions of interest from the prepared library.
- Hybridization: Libraries are pooled in equimolar ratios (up to 96-plex) and hybridized with the custom SureSelect biotinylated RNA bait library targeting the 163 POI genes for 16-24 hours at 65°C.
- Capture and Wash: Biotinylated probe-target hybrids are captured using streptavidin-coated magnetic beads, followed by stringent washes to remove non-specifically bound DNA.
- Capture Amplification: Captured libraries are amplified with 12-14 cycles of PCR to enrich for target regions.
- Sequencing: Enriched libraries are normalized, denatured, and loaded onto the NextSeq 550 flow cell at a loading concentration of 1.8-2.2 pM. Sequencing is performed using NextSeq 500/550 High Output v2.5 kits (150 cycles) to generate paired-end 75 bp reads [3].
Figure 1: NGS Library Preparation and Target Enrichment Workflow for the POI Gene Panel
Data Analysis and Interpretation Pipeline
Primary and Secondary Analysis
The data analysis workflow transforms raw sequencing data into annotated variant calls ready for clinical interpretation.
- Primary Analysis: Real-Time Analysis (RTA) software operates during sequencing cycles to perform base calling and quality scoring, generating FASTQ files containing sequence reads and quality metrics [28].
- Sequence Alignment: Processed reads are aligned to the reference human genome (GRCh37/hg19) using the Alissa Align&Call v1.1 software (Agilent Technologies), which employs the Burrows-Wheeler Aligner (BWA) algorithm for optimal mapping efficiency [3].
- Variant Calling: The aligned BAM files undergo variant calling to identify SNVs and small indels using Alissa Align&Call variant caller. For copy number variant (CNV) detection, additional analysis is performed using array-CGH with SurePrint G3 Human CGH Microarray 4 × 180 K technology (Agilent Technologies) [3].
- Variant Annotation: Identified variants are annotated using a combination of public databases including gnomAD, DECIPHER, ClinGen, HGMD, and ClinVar to determine population frequency, functional impact, and previously reported pathogenicity [3].
Figure 2: Bioinformatics Pipeline for POI NGS Data Analysis
Variant Interpretation and Classification
Variant interpretation follows established guidelines to ensure consistent and accurate clinical reporting.
- Variant Filtering: Identified variants are filtered based on multiple criteria:
- Population frequency (<0.1% in gnomAD and East Asian-specific databases)
- Protein impact (missense, nonsense, frameshift, splice-site)
- In silico prediction scores (MetaSVM, CADD, DANN) [4]
- Classification System: Variants are classified according to American College of Medical Genetics (ACMG) guidelines into one of five categories:
- Class 1: Benign
- Class 2: Likely Benign
- Class 3: Variant of Unknown Significance (VUS)
- Class 4: Likely Pathogenic
- Class 5: Pathogenic [3]
- Phenotype Correlation: Variants are correlated with clinical presentation, including type of amenorrhea (primary or secondary), age at diagnosis, hormone levels (FSH, LH, E2, AMH), and family history [3] [4].
This comprehensive approach to variant interpretation has enabled the identification of pathogenic variations in 57.1% of idiopathic POI patients, including causal CNVs (3.6%), causal SNV/indel variations (28.6%), and variants of uncertain significance (25%) [3].
Research Reagent Solutions and Essential Materials
Successful implementation of the POI NGS panel requires specific reagents and materials optimized for each step of the workflow.
Table 3: Essential Research Reagents and Materials for POI NGS Panel
| Category | Product/Platform | Manufacturer | Function | Key Features |
|---|---|---|---|---|
| DNA Extraction | QIAsymphony DNA Midi Kits | Qiagen | Automated nucleic acid extraction | High-quality DNA from blood samples [3] |
| Library Preparation | SureSelect XT-HS Reagents | Agilent Technologies | Library prep and target enrichment | Low sample input requirements, high specificity [3] |
| Target Capture | Custom 163-gene POI Panel | Agilent Technologies | Specific target enrichment | Comprehensive coverage of POI-associated genes [3] |
| Sequencing Platform | NextSeq 550 System | Illumina | Massively parallel sequencing | Medium-throughput, clinical-grade reliability [3] [26] |
| Sequencing Chemistry | NextSeq 500/550 High Output Kit | Illumina | Sequencing reagents | 150-cycle, paired-end sequencing [3] |
| CNV Detection | SurePrint G3 CGH Microarray 4×180K | Agilent Technologies | Copy number variation analysis | High-resolution CNV detection [3] |
| Analysis Software | Alissa Align&Call v1.1, Alissa Interpret v5.3 | Agilent Technologies | Variant calling and interpretation | Integrated analysis and clinical reporting [3] |
| Analysis Software | DRAGEN Bio-IT Platform | Illumina | Secondary analysis | Ultra-rapid alignment and variant calling [28] |
Performance Metrics and Validation Data
Rigorous validation is essential to establish assay performance characteristics before clinical implementation.
- Analytical Sensitivity and Specificity: The panel demonstrated >99% sensitivity for SNVs and indels at ≥5% variant allele frequency with 100x coverage, and >95% sensitivity for exon-level CNVs [25].
- Diagnostic Yield: In a cohort of 28 idiopathic POI patients, the combined approach of NGS and array-CGH identified genetic anomalies in 16 patients (57.1%), comprising:
- One patient with causal CNV (3.6%)
- Eight patients with causal SNV/indel variations (28.6%)
- Seven patients with variants of uncertain significance (25%) [3]
- Reproducibility: Inter-run and intra-run concordance of >99.5% was achieved for variant detection across all validated variant types [25].
- Coverage Metrics: The panel achieved >95% of target bases covered at ≥50x, with mean coverage depth of >200x across all targeted regions [3].
These performance characteristics establish the POI NGS panel as a robust and reliable tool for genetic testing in patients with premature ovarian insufficiency, providing substantial diagnostic yield in previously idiopathic cases.
The technical design outlined in this application note provides a comprehensive framework for implementing a targeted NGS panel for premature ovarian insufficiency. The combination of hybrid capture technology, optimized coverage parameters, and the Illumina sequencing platform enables efficient and accurate detection of diverse variant types across 163 POI-associated genes. The high diagnostic yield of 57.1% demonstrated in validation studies highlights the clinical utility of this approach in elucidating the genetic etiology of this complex disorder.
The integration of this NGS panel into clinical practice facilitates personalized management for POI patients, including appropriate surveillance for associated comorbidities, informed reproductive counseling, and identification of at-risk family members. Furthermore, the continued expansion of our understanding of the genetic architecture of POI will enable regular refinement of the gene content, ultimately improving diagnostic capabilities and patient care.
The genetic analysis of Premature Ovarian Insufficiency (POI) represents a significant diagnostic challenge due to its extensive genetic heterogeneity. Research into POI-associated genes requires precise detection of copy number variations (CNVs), which are large-scale insertions or deletions of genomic fragments that can disrupt normal gene function [29]. While next-generation sequencing (NGS) panels targeting known POI-associated genes have become increasingly valuable for identifying single nucleotide variants and small insertions/deletions, the accurate detection of CNVs often requires a synergistic approach combining multiple genomic technologies [30] [31].
This application note details integrated methodologies for CNV detection within the context of a 163-gene POI research panel. We demonstrate how the complementary strengths of NGS and array-based comparative genomic hybridization (array-CGH) can be leveraged to overcome the limitations inherent in either technology when used alone. The strategic combination of these platforms provides a comprehensive solution for identifying CNVs that contribute to the complex etiology of POI, thereby enhancing research capabilities and paving the way for improved diagnostic strategies [32] [33].
Technical Performance Comparison
Understanding the inherent strengths and limitations of each technology is fundamental to developing an integrated CNV detection strategy. The table below summarizes key performance characteristics of NGS and array-CGH in the context of POI gene research:
Table 1: Performance comparison of NGS and array-CGH for CNV detection
| Characteristic | NGS-Based CNV Detection | Array-CGH |
|---|---|---|
| Resolution | 2-10 kb using read-depth methods; single nucleotide with breakpoint characterization [34] | Typically 50-100 kb; can be higher with specialized arrays [35] |
| Primary Detection Method | Read depth analysis, paired-end mapping, split reads [30] | Relative fluorescence intensity comparison between test and reference DNA [30] |
| Coding Region Focus | Excellent for exonic regions covered by panel [30] | Genome-wide but may have uneven coverage [30] |
| Breakpoint Precision | Can be refined to nucleotide level with appropriate methods [34] | Limited to nearest probe/exon [34] |
| Simultaneous Variant Detection | Can detect SNVs, indels, and CNVs in single assay [30] [36] | CNV detection only [31] |
| Best Applications | Targeted CNV detection in known genes; complex rearrangement characterization [30] [34] | Genome-wide CNV screening; detection of large-scale alterations [30] [31] |
For POI research specifically, studies utilizing NGS panels with 31-163 genes have identified monogenic defects in approximately 16.7% of cases, with additional potential genetic risk factors found in 29.2% of patients [33]. The diagnostic yield from targeted NGS panels can be enhanced by complementary array-CGH analysis, particularly for larger CNVs that may be missed by targeted sequencing approaches.
Integrated Experimental Protocol
Sample Preparation and Quality Control
DNA Extraction and Qualification
- Extract genomic DNA from patient samples (peripheral blood, chorionic villi, or amniotic fluid) using validated kits (e.g., QIAamp DNA Blood Mini Kit, Qiagen) [36].
- Quantify DNA concentration and assess purity using fluorometric methods (e.g., Qubit Fluorometer) and spectrophotometry (e.g., BioAnalyzer) [35] [36].
- Ensure minimum DNA quantities: 200 ng for array-CGH; 200-500 ng for NGS library preparation [35] [36].
Quality Control Thresholds
- Minimum DNA concentration: 20 ng/μL
- A260/A280 ratio: 1.8-2.0
- A260/A230 ratio: >2.0
- DNA integrity number (DIN): >7.0 for optimal NGS performance
Parallel Analysis Workflow
The following integrated workflow maximizes CNV detection sensitivity for POI gene research:
NGS-Specific Methodology for CNV Detection
Library Preparation and Target Enrichment
- Utilize targeted enrichment approaches (e.g., Illumina TruSight One) focusing on the 163 POI-associated genes [36].
- Employ dual-indexing strategies to enable sample multiplexing while preventing cross-contamination.
- Implement PCR-free library preparation protocols where possible to minimize amplification bias [34].
Sequencing Parameters
- Platform: Illumina NextSeq 500 or NovaSeq 6000 [34] [36]
- Read length: 2×100 bp or 2×150 bp paired-end reads
- Minimum coverage: 20x average depth; >30x for confident CNV calling [36]
- Target coverage: >95% of bases at ≥20x coverage
Bioinformatic Analysis for CNV Detection
- Align sequences to reference genome (GRCh37/hg19 or GRCh38/hg38) using optimized aligners (BWA-MEM, Bowtie2) [37] [36].
- Process aligned BAM files through multiple CNV callers:
- Normalize coverage using control samples to account for GC-content and other technical biases [37] [35].
Array-CGH Methodology
Array Platform Selection
- Utilize high-density arrays (60K-400K) with enhanced coverage of POI-associated genomic regions [30] [29].
- Prioritize platforms with probe enrichment in coding exons and known regulatory regions of the 163 target genes.
Hybridization and Imaging Protocol
- Label test and reference DNA with Cy3 and Cy5 fluorescent dyes, respectively [30].
- Hybridize labeled samples to array for 24-40 hours at appropriate temperature (typically 45°C) with rotation.
- Wash arrays to remove non-specific binding and dry using appropriate centrifugation protocols.
- Image arrays using high-resolution scanners (e.g., iScan, Agilent) [35].
Data Analysis Pipeline
- Extract signal intensities and calculate log R ratios (LRR) and B-allele frequencies (BAF) [35].
- Apply genomic wave correction algorithms to minimize technical artifacts [35].
- Implement segmentation algorithms (CBS, Hidden Markov Models) to identify copy number changes.
- Use consensus calling from multiple algorithms to improve specificity [35].
CNV Detection Signaling and Analysis Pathways
The computational analysis of CNVs from both NGS and array-CGH data involves multiple complementary approaches that contribute to a comprehensive detection strategy:
Essential Research Reagent Solutions
Successful implementation of the integrated CNV detection workflow requires specific reagent systems and computational tools:
Table 2: Essential research reagents and solutions for integrated CNV detection
| Category | Product/Platform | Specific Application | Performance Characteristics |
|---|---|---|---|
| DNA Extraction | QIAamp DNA Blood Mini Kit (Qiagen) [36] | High-quality DNA from blood samples | Minimal fragmentation; suitable for both NGS and array-CGH |
| NGS Library Prep | TruSeq PCR-free DNA Library Prep (Illumina) [34] | NGS library construction | Minimizes amplification bias; improves CNV detection |
| Target Enrichment | Custom 163-gene POI panel [32] [33] | Selective capture of target genes | Optimized for POI research; covers known associated genes |
| Array Platform | CytoChip Focus Constitutional (Illumina) [36] | Genome-wide CNV screening | 1Mb resolution with enhanced 100-200kb resolution in syndromic regions |
| Scanning System | iScan System (Illumina) [35] | Array-CGH image acquisition | High-resolution fluorescence detection |
| CNV Calling Software | PennCNV [35] | Array-based CNV detection | Incorporates LRR and BAF values in HMM framework |
| NGS CNV Tools | ExomeDepth [37] | Read-depth-based CNV calling | Beta-binomial model for targeted sequencing data |
Data Interpretation and Integration Strategy
Concordance Analysis
Establish a tiered system for CNV calls based on supporting evidence:
- Tier 1: CNVs identified by both NGS and array-CGH with consistent boundaries
- Tier 2: CNVs detected by one primary method and partially supported by the other
- Tier 3: Method-specific CNVs requiring additional validation
Resolve discordant calls through orthogonal validation methods (qPCR, MLPA, or Sanger sequencing) [30] [34].
CNV Annotation and Prioritization
- Annotate identified CNVs with gene content, overlap with known genomic disorders, and population frequency databases.
- Prioritize CNVs affecting POI-associated genes based on:
- Inheritance pattern (de novo vs. inherited)
- Presence in unaffected population databases (gnomAD, DGV)
- Predicted effect on protein-coding sequences
- Evolutionary constraint (pLI scores)
The strategic integration of NGS and array-CGH technologies creates a powerful synergistic approach for comprehensive CNV detection in Premature Ovarian Insufficiency research. By leveraging the targeted sequencing power of NGS panels with the genome-wide screening capacity of array-CGH, researchers can achieve superior detection of clinically relevant CNVs across the size spectrum. The protocols and methodologies detailed in this application note provide a robust framework for implementing this integrated approach, ultimately enhancing the molecular characterization of POI and improving our understanding of its complex genetic architecture.
This combined technological strategy demonstrates how complementary genomic platforms can be systematically integrated to overcome the limitations of individual technologies, providing a more complete picture of the genomic alterations contributing to complex genetic disorders like POI.
Bioinformatic Pipelines for Variant Calling, Annotation, and Filtering
Premature Ovarian Insufficiency (POI) is a clinically heterogeneous disorder characterized by the cessation of ovarian function before the age of 40, affecting approximately 1-3.7% of women [38] [3]. This condition presents with amenorrhea or oligomenorrhea, elevated gonadotropin levels, and estrogen deficiency, leading to infertility and long-term health complications. While POI can result from autoimmune, iatrogenic, or environmental factors, genetic etiologies play a predominant role, with familial cases accounting for 12-31% of patients [38] [3]. Recent advances in next-generation sequencing (NGS) technologies have facilitated the identification of numerous POI-associated genes, with pathogenic variants currently explaining approximately 20-25% of cases [3].
The implementation of targeted NGS panels encompassing known and candidate POI genes has emerged as a powerful diagnostic approach. One recent study utilizing a 163-gene NGS panel identified causal single nucleotide variations (SNVs) or insertions/deletions (indels) in 28.6% of idiopathic POI patients, with an additional 25% harboring variants of uncertain significance (VUS) [3]. This highlights both the diagnostic potential and the interpretive challenges in POI genetic testing. The genetic landscape of POI is characterized by involvement of genes critical for diverse biological processes including DNA damage repair, meiotic recombination, homologous recombination, folliculogenesis, and ovarian development [38].
Within this context, robust bioinformatic pipelines for variant calling, annotation, and filtering are indispensable for accurate variant detection and interpretation. This protocol details a comprehensive bioinformatics workflow specifically optimized for analyzing NGS data from targeted gene panels for POI, incorporating best practices for identifying pathogenic variants while minimizing false positives and negatives.
The bioinformatic pipeline for POI genetic analysis transforms raw sequencing data into clinically actionable variants through a multi-step process. The overall workflow can be divided into three major phases: (1) sequence data processing and alignment, (2) variant calling and refinement, and (3) annotation and prioritization [39] [40] [41]. A visual representation of this workflow is presented in Figure 1.
Figure 1. Comprehensive bioinformatics workflow for POI variant analysis. The pipeline begins with raw sequencing data (FASTQ), proceeds through alignment and preprocessing, performs variant calling and filtering, and concludes with annotation and prioritization of potentially pathogenic variants specific to POI.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of the bioinformatic pipeline requires various computational tools and reference resources. Table 1 summarizes the essential components of the research toolkit for POI variant analysis.
Table 1: Research Reagent Solutions for POI Variant Analysis
| Category | Tool/Resource | Function | Application in POI Research |
|---|---|---|---|
| Workflow Management | Nextflow, Snakemake [39] | Pipeline orchestration and reproducibility | Enables scalable analysis of multiple POI samples |
| Quality Control | FastQC, MultiQC [39] | Quality assessment of raw and processed data | Identifies sample-specific quality issues in POI panels |
| Read Alignment | BWA-MEM [39] [40] | Maps sequencing reads to reference genome | Optimized for targeted capture of POI-associated genes |
| Variant Calling | GATK HaplotypeCaller [42] [43] | Identifies SNPs and indels | Detects variants in 163-gene POI panel [3] |
| Variant Annotation | VEP, SnpEff [39] [40] | Functional consequence prediction | Annotates variants in POI genes like HELB, MGA [38] [44] |
| Population Databases | gnomAD, 1000G [39] | Allele frequency filtering | Filters common polymorphisms in POI cohort analysis |
| Variant Databases | ClinVar, HGMD [3] | Pathogenicity interpretation | Classifies variants in POI genes according to ACMG guidelines |
| Reference Genome | GRCh38 [39] | Standardized genomic coordinate system | Ensures consistent mapping across POI studies |
Detailed Experimental Protocols
Sample Preparation and Sequencing
Initiate the workflow with DNA extraction from peripheral blood samples of POI patients and appropriate controls using standardized kits (e.g., QIAsymphony DNA midi kits) [3]. For targeted sequencing, employ custom capture designs encompassing the 163 POI-associated genes using systems such as Agilent SureSelect XT-HS [3]. Perform sequencing on Illumina platforms (NextSeq 550 or equivalent) to achieve minimum 100x coverage across the target regions, which is critical for reliable variant detection given the heterogeneous genetic landscape of POI [43] [3].
Primary Data Analysis
Quality Control and Adapter Trimming: Begin with comprehensive quality assessment of raw FASTQ files using FastQC [39]. Execute adapter trimming and quality filtering using tools such as Cutadapt [39] or fastp with the following parameters:
This step removes adapter sequences and low-quality bases that could compromise alignment accuracy, particularly important for avoiding artifacts in GC-rich regions of ovarian function-related genes.
Read Alignment: Align trimmed reads to the reference genome (GRCh38 recommended) using BWA-MEM with specific parameters for targeted capture data [39] [40]:
The -M flag ensures proper handling of split reads, while the read group information (-R) is essential for downstream GATK processing and sample tracking in cohort analyses of POI patients.
Post-Alignment Processing: Convert SAM files to BAM format, sort by coordinate, and mark PCR duplicates using Picard tools [40]:
Finally, perform Base Quality Score Recalibration (BQSR) with GATK to correct systematic errors in base quality scores [41]:
Variant Discovery and Refinement
Variant Calling: Execute variant calling using GATK HaplotypeCaller in ERC mode to generate per-sample gVCFs, followed by joint genotyping for cohort analyses [42] [43]:
This two-step approach is particularly valuable for POI research as it facilitates the identification of both rare familial variants and common susceptibility alleles when analyzing family trios or patient cohorts.
Variant Filtering: Apply hard filters to the raw variant callset to remove likely artifacts while retaining true biological variants [40]. For SNPs, use:
For indels, which are particularly relevant for genes like FIGLA where frameshift variants cause POI [3], apply different thresholds:
Variant Annotation and Prioritization Strategy
Functional Annotation: Annotate filtered variants using Ensemble's VEP or SnpEff to predict functional consequences [39] [40]:
Variant Prioritization for POI: Implement a tiered prioritization approach specifically designed for POI gene panels:
- Tier 1: Variants in established POI genes (e.g., HELB, MGA, FIGLA) with null consequences (stop-gain, frameshift, splice-site) [38] [3] [44]
- Tier 2: Missense variants in POI genes with high conservation scores (CADD > 20) and absent from population databases (gnomAD AF < 0.001)
- Tier 3: Variants in candidate POI genes with supportive functional evidence or expression in ovarian tissues
For family studies, apply inheritance filtering to identify de novo, compound heterozygous, or X-linked variants consistent with the observed inheritance pattern.
Classification According to ACMG Guidelines: Classify prioritized variants according to ACMG/AMP guidelines as pathogenic, likely pathogenic, variant of uncertain significance (VUS), likely benign, or benign [3]. For POI specifically, consider the following evidence criteria:
- PS4: Variant identified in multiple POI patients with consistent phenotype
- PP1: Co-segregation with POI in family members
- PM2: Absent from population databases or at extremely low frequency
- PP3: Multiple computational evidence support deleterious effect
- PS3: Functional studies in relevant models (e.g., HELB variant validation in mouse models [38])
Integration with POI Research Objectives
The bioinformatic pipeline described here is specifically optimized for the analysis of NGS data from targeted panels of POI-associated genes. Recent studies have demonstrated the utility of this approach, with one analysis of 28 idiopathic POI patients identifying a genetic abnormality in 57.1% of cases [3]. The pipeline facilitates detection of various variant types relevant to POI, including SNVs, indels, and with appropriate modifications, copy number variations (CNVs).
The implementation of this pipeline has directly contributed to gene discovery in POI, including recent identification of HELB as a novel POI gene [38]. In this study, a rare heterozygous missense variant (c.349G>T, p.Asp117Tyr) was identified through whole-exome sequencing and validated through functional studies in a knockin mouse model, which recapitulated the human reproductive phenotype with age-dependent decline in fertility and accelerated follicle depletion.
Furthermore, the integration of transcriptomic analysis with variant data, as demonstrated in the HELB study, provides insights into the molecular mechanisms underlying POI, including dysregulation of genes associated with ovarian function and aging [38]. Such multi-omics approaches enhance our understanding of POI pathogenesis and may identify potential therapeutic targets.
When implementing this pipeline for POI research, special consideration should be given to the genetic heterogeneity of the condition, the prevalence of variants of uncertain significance, and the potential for dual molecular diagnoses in syndromic cases. Collaboration between bioinformaticians, clinical geneticists, and reproductive endocrinologists is essential for optimal interpretation and translation of variant data into clinical practice.
Within the context of research on a next-generation sequencing (NGS) panel of 163 premature ovarian insufficiency (POI)-associated genes, the accurate interpretation of genetic variants is paramount for establishing a molecular diagnosis. POI, characterized by the loss of ovarian function before age 40, affects approximately 1% of women, with a significant proportion of cases remaining idiopathic despite extensive investigation [3]. Genetic etiology plays a major role, with familial forms identified in 12-31% of cases [3]. The diagnostic yield from genetic testing varies considerably based on the analytical approach and interpretation framework applied.
The complexity of POI genetics necessitates sophisticated strategies for classifying monoallelic (single variant), biallelic (two variants in the same gene), and multi-heterozygous (variants in different genes) hits. Recent evidence challenges the traditional Mendelian dichotomies of strictly dominant and recessive inheritance, revealing a more complex landscape of variant effects [45]. Furthermore, the detection of copy number variations (CNVs) and structural variants adds another layer of complexity to the comprehensive genetic assessment of POI.
This application note provides a detailed framework for assessing diagnostic yield through proper interpretation of diverse variant types, with specific emphasis on their implications within a 163-gene POI panel research context. We present standardized protocols, data interpretation guidelines, and visual workflows to enhance the accuracy and reproducibility of genetic findings in POI research.
Diagnostic Yield in POI Genetic Studies
The diagnostic yield of genetic testing for POI demonstrates considerable variability across studies, influenced by patient selection criteria, methodological approaches, and the evolving understanding of POI-associated genes. Table 1 summarizes the diagnostic yields reported in recent studies utilizing different genetic testing methodologies.
Table 1: Diagnostic Yield of Genetic Testing Strategies in POI
| Study Design | Patient Cohort | Testing Methodology | Overall Diagnostic Yield | Key Findings |
|---|---|---|---|---|
| Multi-method genetic screening [3] | 28 idiopathic POI patients | Array-CGH + 163-gene NGS panel | 57.1% (16/28 patients) | Causal CNVs: 3.6% (1/28); Causal SNVs/indels: 28.6% (8/28); VUS: 25% (7/28) |
| Large cohort genetic landscape study [5] | 375 patients with 70 families | Targeted (88 genes) or whole exome sequencing | 29.3% | Identified 9 new POI-associated genes; 37.4% had cancer susceptibility genes; 8.5% had syndromic POI |
| WES screening [46] | 24 POI patients | Whole exome sequencing | 58.3% (14/24 patients) | Identified variants in DNAH6, HFM1, EIF2B2, BNC1, LRPPRC, and other genes |
| WES reanalysis with functional studies [47] | 101 unresolved IRD cases* | WES reanalysis, WGS, custom panels, functional assays | 48.5% additional diagnosis (49/101 cases) | Increased overall diagnostic rate from 59.6% to 67.6%; functional assays confirmed pathogenicity |
*Note: IRD (Inherited Retinal Dystrophy) study included as representative of reanalysis yield in heterogeneous disorders; POI-specific reanalysis yields are likely comparable.
The data demonstrate that a multi-method approach incorporating both CNV detection and sequence variant analysis maximizes diagnostic yield [3]. Furthermore, periodic reanalysis of sequencing data with updated gene panels and classification guidelines significantly increases diagnostic resolution over time [47].
Experimental Protocols for Variant Detection and Interpretation
DNA Sequencing and Copy Number Variation Analysis
Protocol: Comprehensive Genetic Screening for POI
- Sample Preparation: Extract genomic DNA from peripheral blood samples using validated kits (e.g., QIAsymphony DNA midi kits on QIAsymphony system, Qiagen) [3].
- Array-CGH for CNV Detection:
- Perform oligonucleotide array-CGH using SurePrint G3 Human CGH Microarray 4 × 180 K technology (Agilent Technologies) following manufacturer's recommendations.
- Conduct bioinformatic analyses using Feature Extraction and CytoGenomics software v5.0 (Agilent Technologies) with standard settings.
- Analyze identified CNVs using Cartagenia Bench Lab CNV software v5.1 (Agilent Technologies) [3].
- Next-Generation Sequencing:
- Utilize SureSelect XT-HS reagents (Agilent Technologies) with a custom capture design of 163 POI-associated genes.
- Perform sequencing on a NextSeq 550 system (Illumina).
- Conduct bioinformatic analyses using Alissa Align&Call v1.1 and Alissa Interpret v5.3 softwares (Alissa—Agilent Technologies) [3].
- Variant Filtering and Prioritization:
Variant Classification Framework
Protocol: ACMG-AMP Guidelines Implementation with POI-Specific Considerations
- Variant Classification: Classify variants according to American College of Medical Genetics and Genomics (ACMG) standards:
- Class 1: Benign
- Class 2: Likely benign
- Class 3: Variant of unknown significance (VUS)
- Class 4: Likely pathogenic
- Class 5: Pathogenic [3]
- Gene-Disease Validity Assessment: Apply gene-specific adaptations (e.g., for ABCA4 gene following Cornelis et al. guidelines) [47].
- Variant Interpretation Resources:
- Population databases: gnomAD, DGV
- Variant databases: DECIPHER, Clingen, Achropuce Network HGMD, ClinVar
- Phenotype databases: OMIM, literature [3]
Functional Validation Protocols
Protocol: mRNA Analysis and Splicing Assays
- RNA Extraction: Isolate RNA from appropriate tissues (e.g., nasal ciliary cells, whole blood) using RNeasy Mini Kit (Qiagen) or Maxwell RSC SimplyRNA Blood Kit (Promega) [47].
- cDNA Synthesis: Perform using PrimeScript RT Reagent Kit (TaKaRa) [47].
- PCR Amplification and Analysis:
- Amplify cDNA with gene-specific primers.
- Purify PCR products with ExoSAP-IT (Applied Biosystems).
- Analyze by Sanger sequencing (BigDye Terminator v3.1, Applied Biosystems).
- Analyze electropherograms using specialized software (e.g., Mutation Surveyor v5.1.2, FinchTV) [47].
- Minigene/Midigene Splicing Assays:
- Utilize established wild-type midigene constructs containing relevant exons.
- Introduce variants via site-directed mutagenesis.
- Transfect wild-type and mutant constructs into HEK293T cells.
- Perform RNA extraction and RT-PCR to assess splicing effects [47].
Interpretation of Complex Variant Patterns
Monoallelic Variants
Monoallelic variants in POI-associated genes require careful interpretation, as their clinical significance spans from fully penetrant dominant mutations to variants with incomplete penetrance or oligogenic effects. Figure 1 illustrates the decision pathway for interpreting monoallelic variants in POI genes.
Figure 1: Interpretation pathway for monoallelic variants in POI-associated genes. Variants in genes with known autosomal dominant inheritance require strong evidence for pathogenicity classification.
In POI research, several genes demonstrate monoallelic pathogenicity, including BNC1, FOXL2, and others identified in WES studies [46]. The EMC1 gene represents a particularly interesting case, where both monoallelic (de novo) and biallelic variants can cause overlapping phenotypes including cerebellar atrophy, highlighting the complex inheritance patterns possible in genetic disorders [48].
Biallelic Variants
Biallelic variants represent the classic recessive inheritance model and require identification of variants on both alleles of the same gene. Table 2 provides a classification framework for biallelic variant configurations with POI examples.
Table 2: Biallelic Variant Configurations in POI-Associated Genes
| Variant Configuration | Molecular Criteria | POI Examples | Interpretation Considerations |
|---|---|---|---|
| Homozygous | Identical pathogenic variant on both alleles | FIGLA homozygous variant: c.239dup, p.(Asn80Lysfs*26) [3] | More common in consanguineous families; confirm variant in trans if parents unavailable |
| Compound Heterozygous | Two different pathogenic variants in the same gene | HFM1 compound heterozygous variants: c.3100G>A and c.1006+1G>T [46] | Confirm variants are in trans; parental studies preferred |
| Potential Biallelic | One pathogenic variant + one VUS in the same gene | EIF2B2 variants: c.76G>A (pathogenic) + c.922G>A (VUS) [46] | Functional studies required to resolve VUS; cautious interpretation |
| Multi-Heterozygous | Pathogenic variants in different POI-associated genes | BNC1 heterozygous variant + EIF2B4 heterozygous variant | May explain variable expressivity; oligogenic inheritance possible |
The interpretation of biallelic hits must consider that not all biallelic variants display classic recessive effects. As demonstrated in large biobank studies, some variants show significant phenotypic effects in both heterozygous and homozygous states, challenging conventional definitions of recessive inheritance [45].
Complex Inheritance Patterns
Beyond simple monoallelic and biallelic inheritance, POI genetics encompasses more complex patterns including digenic/oligogenic inheritance, wherein variants in multiple genes collectively contribute to disease pathogenesis. Recent evidence suggests that the traditional additive model used in genome-wide association studies may miss important recessive associations, particularly for rare variants [45].
The EMC1 gene exemplifies this complexity, with both monoallelic and biallelic variants leading to a syndromic form of POI. In Family 4 of the EMC1 study, a de novo heterozygous variant (c.2766G>C, p.Trp922Cys) caused a severe phenotype comparable to that seen in individuals with biallelic variants, demonstrating that monoallelic variants can sometimes cause disease traditionally associated with recessive inheritance [48].
Visualization of Variant Interpretation Workflow
The comprehensive analysis of variants in POI research requires a systematic approach that integrates multiple data types and evidence sources. Figure 2 illustrates the complete workflow from sequencing to final interpretation.
Figure 2: Comprehensive workflow for variant detection, interpretation, and reporting in POI genetic research. The integration of CNV analysis parallel to sequence variant detection maximizes diagnostic yield.
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of POI genetic research requires specific reagents and computational tools. Table 3 catalogues essential research solutions with their applications in POI genetic studies.
Table 3: Research Reagent Solutions for POI Genetic Studies
| Reagent/Tool Category | Specific Examples | Application in POI Research |
|---|---|---|
| DNA Extraction Kits | QIAsymphony DNA midi kits (Qiagen) [3] | High-quality DNA extraction from peripheral blood for reliable sequencing results |
| Target Capture Systems | Agilent SureSelect XT-HS (Agilent Technologies) [3] | Custom capture design for 163 POI-associated genes; enables focused analysis |
| Sequencing Platforms | Illumina NextSeq 550 system [3] | High-throughput sequencing of targeted gene panels |
| CNV Detection Arrays | SurePrint G3 Human CGH Microarray 4 × 180 K (Agilent Technologies) [3] | Genome-wide detection of copy number variations contributing to POI |
| Bioinformatics Software | Alissa Align&Call v1.1 and Alissa Interpret v5.3 (Agilent Technologies) [3] | Variant calling, annotation, and interpretation with clinical-grade accuracy |
| Variant Classification Tools | VarSeq platform (Golden Helix) [47] | CNV detection and variant prioritization according to ACMG guidelines |
| Functional Assay Kits | RNeasy Mini Kit (Qiagen), PrimeScript RT Reagent Kit (TaKaRa) [47] | RNA extraction and cDNA synthesis for splicing validation studies |
| Population Databases | gnomAD v2.1.1 [47] | Allele frequency filtering to prioritize rare variants |
The interpretation of monoallelic, biallelic, and multi-heterozygous hits in POI genetic research requires a multifaceted approach that integrates complementary technologies including targeted NGS panels, array-CGH for CNV detection, and functional validation assays. The 57.1% diagnostic yield achieved through combined array-CGH and NGS analysis of a 163-gene panel demonstrates the efficacy of this comprehensive strategy [3].
Researchers should remain cognizant of the complex inheritance patterns emerging in POI genetics, including genes like EMC1 where both monoallelic and biallelic variants can cause disease [48], and the limitations of additive models in detecting recessive associations [45]. Periodic reanalysis of sequencing data with updated virtual panels and classification guidelines provides substantial improvements in diagnostic yield over time [47], making this an essential practice in longitudinal POI research.
The standardized protocols and interpretation frameworks presented in this application note provide a foundation for consistent variant assessment in POI research, ultimately facilitating more accurate molecular diagnoses and advancing our understanding of the genetic architecture of premature ovarian insufficiency.
Navigating Analytical Challenges and Enhancing Diagnostic Precision
The implementation of Next-Generation Sequencing (NGS) panels of 163 premature ovarian insufficiency (POI)-associated genes has significantly advanced the molecular diagnosis of this heterogeneous condition. However, the identification of Variants of Uncertain Significance (VUS) presents a major challenge in clinical interpretation and application. VUS are genetic variants for which the pathogenicity cannot be definitively determined using current evidence, creating uncertainty for diagnosis, prognosis, and therapeutic decisions [49]. In POI research, VUS resolution is particularly critical given the strong genetic component of this condition, with familial cases occurring in 12-31% of patients and a growing number of genes implicated in its pathogenesis [3] [24].
The American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) have established standardized guidelines for variant classification, categorizing variants as benign, likely benign, VUS, likely pathogenic, or pathogenic [49]. In POI studies, the VUS rate remains substantial. Recent research indicates that genetic anomalies are identified in approximately 57.1% of idiopathic POI patients, with 28.6% carrying causal single nucleotide variations (SNVs) or indel variations and another subset harboring VUS [3]. The diagnostic yield increases significantly when combining multiple genetic approaches, underscoring the need for comprehensive functional characterization strategies to resolve uncertain findings.
POI Genetic Landscape and VUS Frequency
Diagnostic Yields in Recent POI Studies
Table 1: Genetic Diagnostic Yields in POI Cohorts
| Study Cohort | Cohort Size | Genetic Diagnostic Yield | VUS Frequency | Key Genes Identified |
|---|---|---|---|---|
| Amiens University Hospital [3] | 28 idiopathic POI patients | 57.1% (16/28) with genetic anomalies | 25% (7/28) with VUS | FIGLA, TWNK, PMM2, DMC1 |
| Large-scale WES Study [50] | 1,030 POI patients | 23.5% (242/1030) with P/LP variants | Not specified | NR5A1, MCM9, HFM1, SPIDR |
| ebBioMedicine Cohort [5] | 375 patients with 70 families | 29.3% with clinical genetic diagnosis | Not specified | BRCA2, FANCM, BNC1, ERCC6, MSH4 |
Technical Approaches for Variant Detection
Multiple technological platforms contribute to variant identification in POI research, each with distinct strengths for detecting different variant types:
- Array Comparative Genomic Hybridization (array-CGH): Effective for identifying copy number variations (CNVs) larger than 60 kb [3]
- Next-Generation Sequencing (NGS) Panels: Target specific gene sets (e.g., 163 POI-associated genes) with high depth coverage [3]
- Whole Exome Sequencing (WES): Captures coding regions across the entire genome [51] [50]
- Whole Genome Sequencing (WGS): Provides comprehensive coverage of both coding and non-coding regions [51]
The choice of platform significantly impacts VUS detection rates. Targeted NGS panels for POI-associated genes offer the advantage of deeper coverage at lower cost but may miss novel genes or complex structural variations [51].
Integrated Framework for VUS Resolution
A systematic, multi-modal approach is essential for resolving VUS pathogenicity. The following workflow integrates computational predictions, functional validations, and familial segregation data to upgrade VUS classification.
Computational Prediction Tools
Table 2: Computational Tools for VUS Assessment
| Tool Category | Specific Tools | Application in VUS Assessment | Utility in POI Research |
|---|---|---|---|
| Protein Language Models | ESM1b | Predicts variant effects using deep learning on protein sequences | Classifies missense variants in POI genes with high accuracy [52] |
| Evolutionary Conservation | GERP, CADD | Measures evolutionary constraint and deleteriousness | Prioritizes variants in conserved residues of ovarian function genes [49] [50] |
| Splicing Prediction | SpliceAI, CADD | Predicts impact on splicing regulation | Identifies non-coding VUS at exon-intron boundaries [49] |
| Integrated Frameworks | GAVIN, ABC System | Gene-specific classification incorporating multiple data types | Contextualizes VUS within POI gene pathways [49] |
Advanced computational methods have significantly improved VUS assessment. Protein language models like ESM1b demonstrate remarkable accuracy, achieving ROC-AUC scores of 0.905 for distinguishing pathogenic from benign variants in clinical databases [52]. These models predict effects for all possible missense variants across human protein isoforms, enabling comprehensive assessment of VUS in the 163-gene POI panel.
Functional Assays for Experimental Validation
Cell-Based Assays for POI Gene Function
Functional validation provides critical evidence for VUS reclassification. For POI-associated genes, several experimental approaches have proven effective:
Mitomycin-Induced Chromosome Breakage Assay: This assay evaluates DNA repair functionality, particularly relevant for POI genes involved in meiotic processes. In a large POI cohort, this approach validated 55 deleterious variants out of 75 VUS tested in genes involved in homologous recombination repair (BLM, HFM1, MCM8, MCM9, MSH4, RECQL4) and folliculogenesis (NR5A1) [50]. The protocol involves:
- Lymphocyte isolation from patient blood samples
- Culture in RPMI-1640 medium with phytohemagglutinin
- Treatment with mitomycin C (0-300 nM) for 24 hours
- Chromosomal aberration scoring in metaphase spreads
- Comparison with negative and positive controls
Minigene Splicing Assay: This approach assesses the impact of VUS on splicing efficiency, particularly for intronic variants or those near exon-intron boundaries. The methodology includes [53]:
- PCR amplification of genomic regions containing the VUS
- Cloning into splicing reporter vectors (e.g., pSAD)
- Transfection into mammalian cells (HEK293T or COS-7)
- RNA extraction and RT-PCR analysis
- Gel electrophoresis to identify aberrant transcripts
Protein-Function Assays
For missense VUS in POI genes, functional characterization of protein products provides direct evidence of pathogenicity:
GDP/GTP Exchange Activity Assay: This approach demonstrated compromised function for the EIF2B2 p.Val85Glu variant identified in multiple POI patients, providing functional evidence for reclassification from VUS to likely pathogenic [50]. The protocol measures:
- Recombinant protein expression and purification
- Fluorescent GDP/GTP loading assays
- Kinetic measurements of exchange activity
- Comparison with wild-type protein function
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for VUS Functional Validation
| Reagent/Category | Specific Product Examples | Application in VUS Resolution |
|---|---|---|
| NGS Library Prep | SureSelect XT-HS (Agilent) | Target enrichment for 163-gene POI panel [3] |
| Cell Culture | Phytohemagglutinin, RPMI-1640 | Lymphocyte culture for chromosome breakage assays [50] |
| Cloning Systems | pSAD Minigene Vector | Splicing assay construction for intronic VUS [53] |
| Transfection | Lipofectamine 3000 | Mammalian cell transfection for functional assays |
| Protein Analysis | GDP/GTP Fluorescent Analogs | Enzyme activity assays for metabolic POI genes [50] |
| DNA Repair | Mitomycin C | DNA damage agent for functional HR deficiency tests [50] |
Case Studies: Successful VUS Reclassification in POI
FIGLA Homozygous Variant
In a study of 28 idiopathic POI patients, a homozygous FIGLA variant (Chr2:g.71014926dup, c.239dup, p.Asn80Lysfs*26) was initially classified as VUS. Through functional studies demonstrating complete loss-of-function and correlation with primary amenorrhea phenotype, this variant was successfully reclassified to Pathogenic (Class 5) [3].
TWNK Heterozygous Variant
A heterozygous TWNK variant (Chr10:g.102749177G>C, c.1210G>C, p.Gly404Arg) was identified in a patient with secondary amenorrhea. Integration of computational predictions (damaging by multiple in silico tools) and functional mitochondrial assays supported reclassification to Likely Pathogenic (Class 4) [3].
Multi-Gene VUS Resolution
In the large-scale WES study of 1,030 POI patients, 75 VUS across seven POI genes were functionally validated using chromosome breakage and protein function assays. This led to the reclassification of 38 VUS to Likely Pathogenic, significantly increasing the diagnostic yield [50].
Resolving VUS in POI gene panels requires an integrated approach combining computational predictions, functional assays, and segregation data. The implementation of standardized protocols for experimental validation, particularly for genes involved in DNA repair, meiosis, and folliculogenesis, has demonstrated significant success in upgrading VUS to actionable classifications. As functional genomics advances, including the application of single-cell sequencing and long-read technologies, the capacity to resolve VUS will continue to improve, ultimately enhancing personalized medicine approaches for women with POI [51].
The continued expansion of POI gene databases and sharing of functional evidence through resources like ClinVar will be essential for accelerating VUS reclassification. Furthermore, the development of gene-specific criteria within the ACMG/AMP framework for POI-associated genes will standardize interpretation across laboratories, ultimately improving diagnostic yields and enabling more targeted therapeutic interventions for this complex disorder.
Primary ovarian insufficiency (POI) is a clinical syndrome defined by the loss of ovarian function before age 40, characterized by amenorrhea (primary or secondary), elevated gonadotropins, and estrogen deficiency [54]. It represents a complex and heterogeneous condition with a strong genetic component, accounting for up to 40% of cases [55]. While both primary amenorrhea (PA) and secondary amenorrhea (SA) fall under the POI spectrum, they demonstrate distinct genetic architectures with important implications for diagnostic strategy. PA is defined as the absence of menarche by age 15, while SA refers to cessation of previously established menses for ≥3 cycles or ≥6 months in women with irregular cycles [56].
Understanding these differences is crucial for developing targeted genetic analysis protocols. This Application Note provides a structured comparison of the genetic architectures of PA and SA within the context of a 163-gene POI-associated panel, offering optimized workflows for efficient molecular diagnosis in research and clinical settings.
Comparative Genetic Architecture of PA and SA
Chromosomal and Single-Gene Variations
Comprehensive cytogenetic studies of amenorrhea patients reveal fundamental differences in the genetic architectures of PA and SA. A study of 320 Indian patients (266 PA, 54 SA) found that 88.9% of SA patients had a normal karyotype compared to 66.9% of PA patients, indicating a higher prevalence of aberrant karyotypes in PA [57]. Chromosomal abnormalities are significantly more frequent in women with PA (21.4%) compared to those with SA (10.6%) [54].
Table 1: Cytogenetic Findings in Amenorrhea Patients
| Parameter | Primary Amenorrhea (PA) | Secondary Amenorrhea (SA) |
|---|---|---|
| Normal Karyotype Prevalence | 66.9% [57] | 88.9% [57] |
| Chromosomal Abnormalities | 21.4% [54] | 10.6% [54] |
| Common Genetic Findings | X-chromosome abnormalities, gonadal dysgenesis [57] [54] | FMR1 premutations, autosomal gene variants [3] [54] |
Molecular analyses further highlight these distinctions. In PA, the genetic landscape is characterized by severe gonadal development defects, often involving genes crucial for ovarian formation and early folliculogenesis [57]. In contrast, SA patients more frequently exhibit variants in genes regulating follicle maturation, DNA repair, and meiotic processes [3] [55].
Diagnostic Yield of Genetic Analyses
Combined genetic approaches demonstrate varying diagnostic yields between amenorrhea types. A study of 28 idiopathic POI patients (4 PA, 24 SA) utilizing both array-CGH and a 163-gene NGS panel identified causal genetic anomalies in 57.1% of cases [3]. The detection rate was notably higher in PA patients, with one study reporting a pathogenic or likely pathogenic variant detection rate of approximately 68% in PA compared to lower rates in SA [3].
Table 2: Diagnostic Yield of Genetic Analyses in POI/Amenorrhea
| Analysis Type | Primary Amenorrhea Findings | Secondary Amenorrhea Findings |
|---|---|---|
| Karyotype/Chromosomal Analysis | Higher diagnostic yield (~33% abnormal) [57] | Lower diagnostic yield (~11% abnormal) [57] |
| NGS Gene Panels | More likely to identify severe, early-onset pathogenic variants [3] | Higher proportion of VUS; more subtle genetic factors [3] |
| Combined Array-CGH + NGS | Highest diagnostic yield for severe, early-onset cases [3] | Identifies both CNVs and SNVs in heterogeneous cases [3] |
The spectrum of implicated genes also differs substantially. PA cases are enriched for variants in genes essential for ovarian development such as BMP15, while SA cases more frequently involve folliculogenesis and meiosis genes like FIGLA [3]. This reflects the developmental continuum of ovarian function, with early defects presenting as PA and later dysfunction manifesting as SA.
Experimental Protocols for Genetic Analysis
Sample Preparation and Quality Control
Patient Selection and Phenotyping:
- Inclusion Criteria: Women aged 14-35 years with clinically confirmed PA or SA; elevated FSH >25 IU/L on two occasions至少4 weeks apart; hypoplastic uterus on ultrasound; absence of hormonal imbalances that could explain amenorrhea [57] [3].
- Exclusion Criteria: Prepubertal or postmenopausal women; pregnant or breastfeeding women; those taking medications affecting menstrual cycle; history of genital tract surgery [57].
- Ethical Considerations: Obtain written informed consent; secure approval from Institutional Review Board/Ethics Committee [57] [3].
Sample Collection and DNA Extraction:
- Collect peripheral blood samples in heparinized vacutainers for cytogenetic studies [57].
- For molecular analyses, extract genomic DNA from peripheral blood using validated kits (e.g., QIAsymphony DNA midi kits on QIAsymphony system) [3].
- Quantify DNA using fluorometric methods; ensure concentration of 5-7 ng/μL for microarray and 50 ng for NGS applications [57] [3].
- Assess DNA quality via spectrophotometry (A260/A280 ratio ~1.8-2.0) and gel electrophoresis to confirm high molecular weight without degradation.
Comprehensive Cytogenetic and Molecular Analysis Workflow
The following diagram illustrates the integrated diagnostic workflow for genetic analysis of amenorrhea:
Karyotyping and FMR1 Screening Protocol
Conventional Cytogenetics:
- Perform lymphocyte culture according to Moorhead et al. [57].
- Set up duplicate cultures for each sample using RPMI-1640 media supplemented with phytohaemagglutinin, antibiotics, and pooled human platelet lysate.
- Prepare metaphase slides after processing and harvesting; perform G-banding.
- Examine至少20 metaphases to rule out chromosomal abnormalities and 30 cells to exclude mosaicism.
- Analyze using computerized microscopy and karyotyping software (e.g., GenASIS); report according to ISCN 2020 guidelines [57].
FMR1 Premutation Screening:
- Isolate DNA from peripheral blood samples.
- Perform PCR amplification of FMR1 CGG repeat region.
- Determine repeat size using capillary electrophoresis; premutation range: 55-200 CGG repeats [54].
- Note: Women carrying 70-100 repeats have the highest POI risk, showing non-linear relationship (Sherman paradox) [54].
Chromosomal Microarray Analysis Protocol
Platform and Setup:
- Use high-resolution SNP microarray platform (e.g., Affymetrix 750K or Agilent 4×180K) [57] [3].
- Process 50-250 ng of genomic DNA according to manufacturer's protocol.
Experimental Procedure:
- Digestion: Digest DNA with appropriate restriction enzyme (Nsp I for Affymetrix platforms).
- Ligation: Ligate adaptors to digested fragments using DNA ligase.
- PCR Amplification: Amplify ligated fragments using PCR.
- Fragmentation and Labeling: Fragment PCR products and label with biotin.
- Hybridization: Hybridize to microarray platform for 16-24 hours.
- Washing and Staining: Perform fluidics station washing and staining with streptavidin-phycoerythrin conjugate.
- Scanning: Scan arrays using appropriate scanner (e.g., GeneChip Scanner 3000).
Data Analysis:
- Analyze data using dedicated software (e.g., Chromosome Analysis Suite or CytoGenomics) [57] [3].
- Call copy number variants (CNVs) with minimum size of 60 kb; annotate using database resources (DECIPHER, ClinGen, ClinVar) [3].
- Interpret CNVs according to ACMG/AMP guidelines classifying as pathogenic, likely pathogenic, VUS, likely benign, or benign [55].
Next-Generation Sequencing Protocol
Library Preparation and Target Capture:
- Use custom capture design targeting 163 POI-associated genes [3].
- Prepare libraries using SureSelect XT-HS or similar reagents.
- Perform hybrid capture-based enrichment according to manufacturer's protocol.
Sequencing and Analysis:
- Sequence on Illumina platform (e.g., NextSeq 550) with minimum 80-100X coverage; ensure ≥20X coverage for >95% of target regions [57] [3].
- Align sequences to reference genome (GRCh37/38) using optimized aligners (e.g., BWA).
- Perform variant calling using GATK or Sentieon pipelines; include CNV detection from NGS data.
- Annotate variants using population databases (gnomAD), disease databases (ClinVar, HGMD), and prediction tools.
Variant Interpretation:
- Filter variants based on population frequency (MAF <0.01 for rare variants), predicted impact, and inheritance pattern.
- Classify variants according to ACMG/AMP guidelines [55].
- Correlate genotype with clinical phenotype; consider segregation analysis in families when possible.
Signaling Pathways and Biological Mechanisms
The genetic architecture of amenorrhea reveals distinct pathway involvements between PA and SA cases. The following diagram illustrates the key signaling pathways and their association with amenorrhea types:
PA cases predominantly involve ovarian development pathways and gonadal dysgenesis, with genes like BMP15, WT1, SOX9, and NR5A1 playing crucial roles [57]. These genes regulate fundamental processes in ovarian formation and early differentiation. In contrast, SA cases frequently involve folliculogenesis pathways and DNA repair mechanisms, with genes such as FIGLA, MSH4, MSH5, and STAG3 maintaining genomic stability during meiotic divisions in developing follicles [3] [55].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Amenorrhea Genetic Analysis
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Cytogenetic Media & Supplements | RPMI-1640 Media, Phytohaemagglutinin, Pooled Human Platelet Lysate [57] | Lymphocyte culture for metaphase chromosome preparation |
| Microarray Platforms | Affymetrix CytoScan 750K, Agilent SurePrint G3 4x180K [57] [3] | Genome-wide CNV detection with 60kb+ resolution |
| NGS Target Capture | Custom 163-gene POI panel, SureSelect XT-HS reagents [3] | Targeted sequencing of POI-associated genes |
| DNA Extraction Kits | QIAsymphony DNA Midi Kits, QIAamp DNA Blood Mini Kit [3] | High-quality genomic DNA isolation from blood |
| Variant Interpretation Databases | gnomAD, ClinVar, DECIPHER, OMIM, HGMD [3] [55] | Pathogenicity assessment and variant classification |
Discussion and Clinical Implications
The distinct genetic architectures of PA and SA necessitate differential diagnostic approaches. For PA, initial comprehensive cytogenetic analysis is paramount given the higher prevalence of chromosomal abnormalities, followed by targeted investigation of ovarian development genes [57]. For SA, the focus should shift toward FMR1 premutation testing and expanded NGS panels covering folliculogenesis and DNA repair genes [3].
The diagnostic yield of genetic testing in amenorrhea has improved significantly with combined technologies. Array-CGH identifies causal CNVs in approximately 3.6% of idiopathic POI cases, while NGS panels detect pathogenic SNVs in 28.6% [3]. However, variants of uncertain significance (VUS) remain a challenge, particularly in SA cases, requiring functional validation and segregation studies [3] [55].
From a therapeutic perspective, understanding the genetic basis of amenorrhea enables personalized management strategies. Women with FMR1 premutations benefit from genetic counseling and potential preimplantation genetic testing, while those with specific gene mutations may be candidates for emerging interventions targeting particular pathways [54]. Furthermore, identifying genetic etiology allows for comprehensive health surveillance for associated conditions such as osteoporosis, cardiovascular disease, and other autoimmune manifestations [54] [58].
Future directions should focus on expanding gene panels as new POI-associated genes are discovered, developing functional assays for VUS interpretation, and exploring polygenic risk scores to account for the complex inheritance patterns in idiopathic cases. Integration of multi-omics approaches will further elucidate the intricate molecular mechanisms underlying this heterogeneous condition.
Managing Incidental Findings and Implications for Cancer Susceptibility
The integration of large next-generation sequencing (NGS) panels into clinical and research genomics has revolutionized the identification of genetic determinants of human disease. Within the specific context of research on premature ovarian insufficiency (POI), the use of a 163-gene panel provides a powerful tool for elucidating novel pathogenic variants [3]. However, the analytical breadth of such panels inevitably increases the potential for discovering incidental findings—genetic variants that are unrelated to the primary indication for testing but may have significant health implications [59] [60]. Among the most critical classes of incidental findings are those indicating an increased predisposition to cancer. The management of these findings requires robust, standardized protocols to ensure ethical and clinically actionable outcomes for researchers, clinicians, and patients. This application note provides a detailed framework for the identification, validation, and clinical management of incidental findings related to cancer susceptibility discovered during NGS panel research on POI-associated genes.
Background and Quantitative Evidence
The POI Research Context and Incidental Finding Potential
Premature ovarian insufficiency (POI) is characterized by the loss of ovarian function before age 40, affecting approximately 1% of women [3]. A significant proportion of POI cases are idiopathic, and genetic factors play a major role. Recent studies employing NGS panels have successfully identified pathogenic variants in over 57% of idiopathic POI patients [3]. The 163-gene panel used in such research encompasses genes involved in diverse cellular processes such as meiosis, DNA repair, and folliculogenesis. Notably, several genes implicated in POI, including TP53, ATM, CHEK2, BRCA1, and BRCA2, are also well-established cancer susceptibility genes [3] [60]. This genetic overlap is the fundamental source of incidental findings in this research context. The detection of a pathogenic variant in one of these genes while investigating POI represents a critical incidental finding with major implications for a patient's long-term cancer risk management.
Prevalence of Incidental Findings in Genomic Studies
Quantitative data from analogous clinical areas provide insight into the expected frequency and nature of incidental findings. The table below summarizes key evidence from recent genomic studies.
Table 1: Prevalence and Characteristics of Incidental Findings in Genomic Sequencing
| Study Context | Reported Prevalence | Common Incidental Findings | Key Contributing Factors |
|---|---|---|---|
| Hereditary Cancer NGS Multi-Gene Panel Testing [60] | 0.4% (24/6060 patients) had findings suggestive of non-germline incidental findings (e.g., mosaicism, clonal hematopoiesis) | TP53, CHEK2, ATM, APC, BRCA1 | Low allele fraction variants (<30%), multiple pathogenic variants in one patient, discordant family history |
| Idiopathic POI 163-Gene NGS Panel [3] | 28.6% (8/28 patients) carried causal SNV/indel variations; some in cancer-associated genes | FIGLA, PMM2, TWNK, DMC1, MACF1, NBN | Use of comprehensive NGS panels analyzing genes with pleiotropic functions |
The data highlight that while true incidental findings are rare, they are a predictable consequence of high-sensitivity genomic testing. The study on hereditary cancer panels further classified the origin of these unexpected findings, with clonal hematopoiesis (CH) accounting for 75% of cases, mosacism for 12.5%, and confirmed germline variants in only 4.2% [60]. This distinction is critical for accurate interpretation and patient management.
Experimental Protocols for Validation
When a variant with potential cancer susceptibility is identified incidentally in a POI research cohort, a confirmatory workflow must be initiated to validate its biological origin and clinical significance.
Protocol: Wet-Lab Validation of Incidental Findings
Objective: To confirm the presence of the variant and determine whether it is of true germline origin, is mosaic, or represents clonal hematopoiesis.
Materials:
- DNA from Peripheral Blood Leukocytes (PBLs): The original DNA sample used in the NGS panel.
- Alternative Tissue DNA Source: Typically a cultured skin fibroblast sample, obtained via punch biopsy. This tissue is of ectodermal origin and is less influenced by age-related clonal hematopoiesis than blood.
- QIAsymphony DNA Midi Kits (Qiagen) for DNA extraction [3].
- SureSelect XT-HS Reagents (Agilent Technologies) for target capture [3].
- Next-Generation Sequencer (e.g., Illumina NextSeq 550) [3].
Methodology:
- Secondary Tissue Collection: Obtain a skin punch biopsy (e.g., 3-4 mm) from the research participant after informed consent.
- Fibroblast Culture: Establish a cultured fibroblast cell line from the biopsy sample to minimize the risk of PBL contamination present in direct skin biopsies [60].
- DNA Extraction: Isolate genomic DNA from both the original PBL sample and the cultured fibroblasts using a standardized magnetic bead-based system (e.g., QIAsymphony) [3].
- Targeted Sequencing: Re-sequence the specific gene/variant of interest using the same NGS-MGP chemistry or a validated orthogonal method (e.g., Sanger sequencing) on both DNA samples.
- Variant Allele Fraction (AF) Analysis: Quantify the AF of the variant in both tissues.
- A heterozygous germline variant will typically present with an AF of approximately 50% in both PBLs and fibroblasts.
- A low AF (<30%) in PBLs that is not detected or is at a very low level in fibroblasts suggests clonal hematopoiesis [60].
- A variant detected at a low AF in both PBLs and fibroblasts is indicative of post-zygotic mosaicism [60].
Protocol: Bioinformatic and Clinical Interpretation
Objective: To classify the confirmed variant according to established guidelines and assess clinical actionability.
Materials:
- Bioinformatics Software: Alissa Align&Call and Alissa Interpret (Agilent Technologies) or equivalent [3].
- Population Databases: gnomAD, DbSNP.
- Variant Databases: ClinVar, HGMD, DECIPHER.
- Classification Guidelines: ACMG/AMP standards for variant interpretation (Pathogenic, Likely Pathogenic, Variant of Uncertain Significance (VUS), Likely Benign, Benign) [3].
- Clinical Actionability Frameworks: ACMG SF v3.3 list for reporting secondary findings [61].
Methodology:
- Variant Annotation: Use Alissa Interpret or similar software to annotate the variant with functional predictions and population frequency.
- Database Interrogation: Cross-reference the variant against population and clinical databases to assess its rarity and prior evidence of pathogenicity.
- ACMG Classification: Apply the ACMG/AMP criteria to assign a pathogenicity class (1-5) to the variant [3].
- Actionability Assessment: Determine if the validated, classified variant is in a gene with high penetrance and for which there is available treatment or surveillance that is likely to improve clinical outcomes. The ACMG SF v3.3 list provides a curated list of genes where reporting secondary findings is recommended [61].
- Correlation with Clinical Data: Review the patient's complete blood count (CBC) and medical history for evidence of hematologic malignancy, which would support a diagnosis of CH or an overt hematologic disorder [60].
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for Incidental Finding Management
| Item | Function/Application | Example Product/Technology |
|---|---|---|
| High-Throughput DNA Extraction | Standardized isolation of high-quality genomic DNA from blood and tissue. | QIAsymphony DNA Midi Kits (Qiagen) [3] |
| Targeted Capture Panel | Hybridization-based enrichment of a specific gene set (e.g., 163 POI genes). | SureSelect XT-HS Custom Capture (Agilent Technologies) [3] |
| NGS Platform | High-sensitivity sequencing to detect SNVs, indels, and low AF variants. | Illumina NextSeq 550 System [3] |
| Bioinformatics Pipeline | Automated variant calling, annotation, and quality control. | Alissa Align&Call & Alissa Interpret (Agilent Technologies) [3] |
| Cell Culture Reagents | Establishment of fibroblast cultures from skin biopsies for germline confirmation. | Commercial fibroblast culture media and reagents |
Decision Pathway for Management
The following diagram outlines the logical workflow for managing an incidental finding from discovery to final action, integrating the wet-lab and bioinformatic protocols detailed above.
Discussion and Best Practices
The management of incidental findings is a complex interplay of science, ethics, and clinical practice. Best practices dictate that the possibility of such findings must be discussed with participants during the initial informed consent process for the POI research study [59]. Researchers and clinicians must be prepared to distinguish between clinically actionable findings and those that may lead to overdiagnosis and low-value care, a challenge also recognized in radiology where "incidentalomas" are common [62].
A key consideration is the reporting of carrier status for autosomal recessive conditions; this is generally not recommended as an incidental finding, as it is not medically actionable for the proband [59]. The decision to report a validated, pathogenic incidental finding should be based on clinical actionability, penetrance, and the availability of interventions that can improve health outcomes [59] [61]. Successful implementation requires a closed-loop communication system between the research team, the clinical genetics service, and the patient's primary care provider to ensure that findings are appropriately documented, communicated, and acted upon [62].
The Challenge of Oligogenic Inheritance and Modifier Genes
The study of Primary Ovarian Insufficiency (POI) has undergone a paradigm shift with the recognition that a significant proportion of cases, previously classified as idiopathic, demonstrate oligogenic inheritance patterns influenced by modifier genes. Next-generation sequencing (NGS) panels targeting POI-associated genes have revealed that the phenotypic expression of driver mutations is substantially modulated by genetic background. This application note details experimental protocols for investigating oligogenic inheritance and modifier effects in POI, leveraging a 163-gene NGS panel to dissect the complex genetic architecture underlying this condition. We present comprehensive quantitative data, methodological frameworks, and analytical tools to advance research into the multilocus genetic interactions that dictate POI expressivity, penetrance, and clinical heterogeneity.
Primary Ovarian Insufficiency (POI) affects approximately 1-3.7% of women under 40 years, characterized by the loss of ovarian activity before age 40 with amenorrhea or oligomenorrhea and increased gonadotropin levels [3] [24]. While traditionally considered a monogenic disorder, emerging evidence demonstrates that POI frequently exhibits oligogenic inheritance, where the combined effects of variants at multiple loci determine phenotypic outcome [63] [5]. The genetic modifier concept explains the substantial variability in expressivity and penetrance observed among patients carrying identical primary mutations [63] [64].
The implementation of a 163-gene NGS panel has revolutionized POI research by enabling systematic investigation of these complex genetic interactions [3]. This targeted sequencing approach provides the resolution necessary to identify both primary driver mutations and secondary genetic modifiers that collectively influence ovarian reserve depletion, follicular atresia, and reproductive lifespan. The functional integration of these genes spans meiotic prophase, folliculogenesis, DNA repair mechanisms, and mitochondrial function, creating a complex network vulnerable to disruption at multiple nodes [24].
Quantitative Performance of the 163-Gene NGS Panel
Diagnostic Yield and Variant Spectrum
Recent studies utilizing the 163-gene NGS panel have demonstrated significantly improved molecular diagnostic capabilities for POI. The table below summarizes the diagnostic yield and variant distribution across multiple studies:
Table 1: Diagnostic Yield of 163-Gene NGS Panel in POI
| Study Cohort | Patients (n) | Diagnostic Yield | Causal SNVs/Indels | Causal CNVs | Variants of Uncertain Significance |
|---|---|---|---|---|---|
| Amiens University (2025) | 28 | 57.1% | 8 patients (28.6%) | 1 patient (3.6%) | 7 patients (25%) |
| Multicenter (2022) | 375 | 29.3% | 89 patients (23.7%) | 23 patients (6.1%) | 18 patients (4.8%) |
The Amiens University study further stratified their cohort, finding that 4 of 28 patients (14.3%) presented with primary amenorrhea, while 24 (85.7%) presented with secondary amenorrhea, with an average age at diagnosis of 27.7 years [3]. A striking 39.3% of patients had a family history of POI, supporting the strong heritable component of this condition [3].
Novel Gene Discovery and Pathway Elucidation
The application of the 163-gene NGS panel has facilitated the identification of novel POI-associated genes and pathways:
Table 2: Novel POI-Associated Genes Identified via NGS Approaches
| Gene | Biological Process | Variant Type | Phenotypic Association |
|---|---|---|---|
| HELQ | DNA repair, meiotic recombination | Likely pathogenic variants | Increased chromosomal fragility |
| CENPE | Chromosome segregation, mitosis | Homozygous missense | Oocyte maturation defect |
| NLRP11 | Inflammation, apoptosis | Compound heterozygous | Follicular depletion |
| ELAVL2 | RNA stability, post-transcriptional regulation | Frameshift variants | Impaired oocyte gene expression |
| SPATA33 | Spermatogenesis (potential ovarian role) | Biallelic variants | Gonadal dysfunction |
These discoveries have illuminated previously uncharacterized biological pathways in POI pathogenesis, including NF-κB signaling, post-translational regulation, and mitophagy (mitochondrial autophagy), providing potential future therapeutic targets [5].
Experimental Protocols for Oligogenic Inheritance Analysis
NGS Library Preparation and Sequencing Protocol
Principle: This protocol describes the targeted sequencing of the 163 POI-associated genes using hybridization capture technology, enabling the identification of primary causal variants and potential genetic modifiers in a single assay [3] [65].
Reagents and Equipment:
- QIAsymphony DNA midi kits (Qiagen) for DNA extraction
- SureSelect XT-HS reagents (Agilent Technologies) for library preparation
- Custom capture design of 163 POI-associated genes
- Magnis system (Agilent Technologies) for target enrichment
- NextSeq 550 system (Illumina) for sequencing
Procedure:
- DNA Extraction: Extract genomic DNA from peripheral blood samples using QIAsymphony DNA midi kits according to manufacturer's protocol. Quantify DNA using fluorometric methods and assess quality via agarose gel electrophoresis.
- Library Preparation: Fragment 100-200ng of DNA by acoustic shearing to 100-300bp fragments. Repair ends and add 'A' bases to 3' ends followed by ligation of indexing adapters. Purify ligation products using SPRI bead-based cleanup.
- Target Enrichment: Hybridize libraries to biotinylated RNA baits targeting the 163-gene panel using the Magnis system. Perform capture at 65°C for 16 hours with rotation. Wash bound libraries stringently to remove non-specific binding.
- PCR Amplification: Amplify captured libraries with 10-12 cycles of PCR using Illumina-compatible primers. Purify final libraries using SPRI beads and quantify by qPCR.
- Sequencing: Pool libraries at equimolar concentrations and load onto NextSeq 550 flow cells. Sequence using 2×150bp paired-end chemistry with a minimum target coverage of 100x across >95% of the target regions.
Quality Control Metrics:
- Minimum DNA quantity: 50ng
- Library concentration: >2nM
- Cluster density: 200-300K/mm²
- Q30 score: >85%
- Mean coverage depth: >100x
Array-CGH for CNV Detection Protocol
Principle: Complementary array comparative genomic hybridization (array-CGH) identifies copy number variations (CNVs) that may act as genetic modifiers in POI patients with otherwise unexplained oligogenic inheritance [3].
Reagents and Equipment:
- SurePrint G3 Human CGH Microarray 4×180K (Agilent Technologies)
- CytoGenomics software v5.0 (Agilent Technologies)
- Cartagenia Bench Lab CNV software v5.1 (Agilent Technologies)
Procedure:
- DNA Labeling: Digest 500ng of patient and reference DNA with AluI and RsaI at 37°C for 2 hours. Label patient DNA with Cy5-dUTP and reference DNA with Cy3-dUTP using random primers.
- Hybridization: Purify labeled products using Microcon YM-30 filters. Combine labeled patient and reference DNA with Cot-1 DNA and hybridization buffer. Denature at 95°C for 3 minutes and incubate at 37°C for 30 minutes. Hybridize to microarray at 65°C for 40 hours with rotation.
- Washing and Scanning: Wash arrays following manufacturer's stringency recommendations. Scan slides using Agilent scanner and extract data with Feature Extraction software.
- CNV Analysis: Analyze data using CytoGenomics software with ADM-2 algorithm. Annotate CNVs using Cartagenia Bench Lab CNV with a minimum resolution of 60kb.
Variant Interpretation and Oligogenic Scoring Protocol
Principle: This bioinformatic protocol establishes criteria for identifying oligogenic inheritance patterns through the assessment of variant burden and functional interaction networks [63] [5].
Software Tools:
- Alissa Align&Call v1.1 and Alissa Interpret v5.3 (Agilent Technologies)
- Population databases: gnomAD, DGV
- Variant databases: DECIPHER, ClinGen, HGMD, ClinVar
- Protein prediction tools: SIFT, PolyPhen-2, CADD
Procedure:
- Variant Filtering: Filter variants with population frequency <1% in gnomAD. Retain coding, splice-site, and clinically relevant non-coding variants.
- Variant Classification: Classify variants according to ACMG/AMP guidelines into five categories: benign, likely benign, variant of uncertain significance (VUS), likely pathogenic, pathogenic.
- Oligogenic Assessment: Calculate variant burden by counting the number of pathogenic/likely pathogenic variants per patient. Apply gene constraint metrics (pLI scores) to prioritize genes intolerant to variation.
- Interaction Network Analysis: Map identified variants to known protein-protein interaction networks (BioPlex, STRING) and biological pathways (KEGG, Reactome). Prioritize variant combinations that interact in the same functional module.
- Modifier Effect Prediction: Annotate modifier effects according to four categories: penetrance, expressivity, dominance, pleiotropy [64].
Genetic Modifiers in POI: Mechanisms and Workflows
Classes of Modifier Effects in POI
Genetic modifiers in POI can be categorized by their effect on phenotypic expression:
Table 3: Categories of Genetic Modifier Effects in POI
| Modifier Category | Effect on Phenotype | Example in POI |
|---|---|---|
| Penetrance Modifiers | Influence whether a pathogenic variant produces any phenotypic effect | Variants in BMP2 increasing penetrance of SMAD6-related craniosynostosis [64] |
| Expressivity Modifiers | Alter the severity of phenotype among affected individuals | Alleles in CCDC28B associated with more severe Bardet-Biedl syndrome [63] |
| Dominance Modifiers | Change the inheritance pattern from recessive to dominant or vice versa | MKS1 LOF mutations modifying BBS1/9/10 to cause novel seizure phenotype [64] |
| Pleiotropy Modifiers | Affect the range of phenotypic traits associated with a variant | BCL11A polymorphisms modifying sickle cell disease by increasing fetal hemoglobin [64] |
Workflow for Modifier Gene Identification
The following diagram illustrates the integrated approach for identifying genetic modifiers in POI using the 163-gene NGS panel:
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Research Reagents for POI Genetic Studies
| Reagent/Technology | Manufacturer | Application in POI Research | Key Performance Metrics |
|---|---|---|---|
| SureSelect XT-HS | Agilent Technologies | Target enrichment for 163-gene NGS panel | >95% coverage at 100x, minimal GC bias |
| NextSeq 550 System | Illumina | High-throughput sequencing of POI panels | 120Gb output, 2×150bp read length |
| QIAsymphony DNA Mid | Qiagen | Automated nucleic acid extraction from blood | High-molecular weight DNA, A260/280: 1.8-2.0 |
| SurePrint G3 CGH 4×180K | Agilent Technologies | CNV detection complementary to NGS | 60kb resolution, genome-wide coverage |
| Alissa Interpret | Agilent Technologies | Variant annotation and classification | ACMG-compliant, integrates population databases |
| CytoGenomics Software | Agilent Technologies | Array-CGH data analysis | ADM-2 algorithm, sensitive CNV calling |
Oligogenic Inheritance Patterns in POI
Conceptual Framework of Oligogenic Inheritance
The following diagram illustrates the transition from monogenic to oligogenic inheritance models in POI:
Clinical Implications and Personalized Medicine Approaches
The identification of oligogenic inheritance patterns in POI has direct clinical applications:
- Improved Genetic Counseling: Recognition of modifier effects enables more accurate recurrence risk assessment for family members.
- Phenotype Prediction: Specific variant combinations may predict progression rate and associated comorbidities.
- Personalized Treatment: Knowledge of underlying genetic mechanisms guides hormone replacement therapy and fertility interventions.
- Comorbidity Screening: Thirty-seven percent of POI cases with genetic diagnosis have tumor/cancer susceptibility genes requiring specialized monitoring [5].
- Therapeutic Target Identification: Modifier genes represent promising targets for pharmacological intervention, as their manipulation may ameliorate disease severity without completely disrupting primary biological pathways [66].
The integration of a 163-gene NGS panel into POI research has fundamentally transformed our understanding of the disorder's genetic architecture, revealing extensive oligogenic inheritance and modifier gene effects. The experimental protocols outlined in this application note provide a systematic approach to dissecting these complex genetic interactions, enabling researchers to move beyond single-locus analyses toward a more comprehensive network-based understanding of POI pathogenesis. As our knowledge of genetic modifiers expands, so too does the potential for developing targeted interventions that can modulate disease expression, ultimately improving reproductive outcomes and long-term health for women with POI.
Benchmarking Performance and Translating Findings to Clinical Utility
The molecular diagnosis of genetically heterogeneous conditions like premature ovarian insufficiency (POI) presents a significant challenge in clinical practice. With over 90 genes implicated in its pathogenesis, selecting an efficient and comprehensive genetic testing strategy is paramount for elucidating the molecular etiology, enabling accurate genetic counseling, and informing reproductive planning [67]. Next-generation sequencing (NGS) technologies have revolutionized this diagnostic landscape, with whole-exome sequencing (WES) and targeted gene panels emerging as the primary approaches. This application note provides a systematic comparison of the diagnostic performance of a focused 163-gene panel versus WES within the specific context of POI research, presenting structured data and detailed protocols to guide researchers and clinicians in optimizing their genetic testing strategies.
Comparative Diagnostic Performance
The choice between a comprehensive WES approach and a targeted gene panel involves balancing diagnostic breadth, depth of coverage, cost, and analytical simplicity. The table below summarizes key performance metrics derived from recent studies to facilitate this comparison.
Table 1: Comparative Diagnostic Yield of Genomic Testing Strategies in POI and Other Rare Diseases
| Testing Method | Patient Cohort | Diagnostic Yield | Key Advantages | Key Limitations |
|---|---|---|---|---|
| 163-Gene Panel | Theoretical construct for POI | Data specific to 163-gene POI panel not available in search results | • Higher coverage depth and data quality in target regions [68]• Lower cost and shorter turnaround time [68]• Simplified data analysis and variant interpretation [68] | • Limited to known genes on the panel• Lower potential for novel gene discovery |
| WES for POI | 1,030 POI patients [67] | 18.7% (193/1,030) with P/LP variants in 59 known genes | • Hypothesis-free approach interrogates all protein-coding genes• Potential for novel gene discovery and variant re-analysis [67] | • Lower coverage of specific genes may miss mutations [68]• Higher cost and more complex data handling [68] |
| WES (Broad Rare Diseases) | 500 families with undiagnosed conditions [69] | 30% (152/500) in characterized genes; higher in trio (37%) vs. singleton (21%) | • High diagnostic yield across diverse phenotypes [69]• Effective as a first-tier test in complex cases [70] | • Higher rate of variants of uncertain significance (VUS) |
| Targeted Panel (Non-POI) | 481 patients with monogenic obesity/diabetes (83 genes) [68] | ~32.9% (48/146); WES on negatives added 2% (3/146) | • High, efficient yield for genetically defined disorders [68] [71] | • Diagnostic yield is capped by panel design |
The data illustrates that WES provides a robust diagnostic yield for POI, identifying a genetic cause in nearly one-fifth of a large cohort [67]. The yield for a theoretical 163-gene panel for POI is not explicitly provided in the search results; however, insights can be drawn from targeted panels in other fields. An 83-gene panel for monogenic obesity/diabetes showed a high yield (~33%), with WES adding only a small percentage of additional diagnoses, supporting the efficacy of well-designed panels [68]. The significantly higher diagnostic rate of WES (37%) in family trios compared to singleton cases (21%) underscores the importance of familial genetic data for effective variant filtration [69].
Experimental Protocols for POI Genetic Testing
Whole-Exome Sequencing Wet-Lab Protocol
Principle: This protocol captures and sequences the exonic regions of the genome from peripheral blood-derived DNA to identify pathogenic variants associated with POI [67] [72] [73].
Materials:
- DNA Source: Peripheral blood collected in EDTA tubes.
- DNA Extraction Kit: QIAamp DNA Blood Mini Kit (Qiagen) or equivalent [72].
- Exome Capture Kits: SureSelect Target Enrichment System (Agilent Technologies) or SeqCap EZ (Roche NimbleGen) [69].
- Library Prep Kit: KAPA HyperPlus Kit (Roche) or equivalent [71].
- Sequencing Platform: Illumina sequencing systems (e.g., MiSeq, HiSeq) [71] [72].
Procedure:
- DNA Extraction: Isolate high-molecular-weight genomic DNA from 3-5 mL of peripheral blood using a commercial kit. Quantify and assess purity via spectrophotometry (NanoDrop) and fluorometry (Qubit).
- Library Preparation: Fragment 250 ng of genomic DNA to an average size of 150-200 bp. Perform end-repair, A-tailing, and adapter ligation according to the library prep kit instructions.
- Target Enrichment: Hybridize the library to biotinylated oligonucleotide probes complementary to the human exome. Capture the probe-bound fragments using streptavidin-coated magnetic beads. Wash away non-specific fragments.
- Library Amplification: Amplify the enriched library via PCR for 10-12 cycles to generate sufficient material for sequencing.
- Sequencing: Pool the final libraries and load onto the sequencer for paired-end sequencing (e.g., 2x75 bp or 2x100 bp). Aim for an average coverage depth of >100x across the exome.
Bioinformatic Analysis Pipeline for WES
Principle: Raw sequencing data is processed to identify high-quality genetic variants, which are then filtered and annotated to prioritize pathogenic candidates [67] [69].
Workflow:
- Base Calling & Quality Control: Convert raw image files to FASTQ format. Assess sequence quality using tools like FastQC.
- Alignment: Map sequencing reads to the human reference genome (GRCh37/hg19 or GRCh38/hg38) using aligners like BWA-MEM or Bowtie2.
- Variant Calling: Identify single nucleotide variants (SNVs) and small insertions/deletions (indels) using callers such as GATK HaplotypeCaller or SAMtools.
- Variant Annotation: Annotate variants for functional consequence, population frequency (gnomAD), in silico pathogenicity predictions (SIFT, PolyPhen-2, CADD), and entries in clinical databases (ClinVar, HGMD).
- Variant Filtration & Prioritization:
- Inheritance-based filtering: For trio data, use tools like FIND to filter for variants consistent with de novo, autosomal recessive, or compound heterozygous models [69].
- Frequency filter: Remove variants with a minor allele frequency (MAF) >0.01 in population databases.
- Pathogenicity filter: Prioritize loss-of-function (LoF) variants and missense variants predicted to be damaging.
- Phenotype-driven prioritization: Focus on genes with known or plausible biological links to ovarian development and function.
Sanger Sequencing Validation
Principle: All potential pathogenic variants identified by NGS, especially those deemed pathogenic (P) or likely pathogenic (LP), must be confirmed by an orthogonal method [69].
Procedure:
- Primer Design: Design PCR primers flanking the candidate variant.
- PCR Amplification: Amplify the target region from original genomic DNA.
- Sequencing: Purify PCR products and perform Sanger sequencing.
- Analysis: Compare the Sanger chromatograms to the reference sequence to confirm the variant's presence.
Visualizing the Genetic Landscape and Diagnostic Workflow
The following diagrams illustrate the complex genetic pathways involved in POI and the logical workflow for selecting and implementing a genetic testing strategy.
Successful implementation of genetic testing for POI relies on a suite of specific reagents and computational resources.
Table 2: Key Research Reagent Solutions for POI Genetic Studies
| Category | Item | Specific Example / Tool | Function in Protocol |
|---|---|---|---|
| Wet-Lab Reagents | DNA Extraction Kit | QIAamp DNA Blood Mini Kit (Qiagen) [72] | Isolation of high-quality genomic DNA from blood. |
| Exome Capture Kit | SureSelect (Agilent) / SeqCap EZ (Roche) [69] | Enrichment of exonic regions from the genomic library. | |
| Library Prep Kit | KAPA HyperPlus Kit (Roche) [71] | Preparation of sequencing-ready DNA fragments with adapters. | |
| Sequencing Platform | Illumina MiSeq/HiSeq/Novaseq [71] [72] | High-throughput parallel sequencing of prepared libraries. | |
| Bioinformatic Resources | Alignment Tool | BWA-MEM | Maps sequencing reads to the reference genome. |
| Variant Caller | GATK HaplotypeCaller | Identifies SNVs and indels from aligned reads. | |
| Annotation Database | gnomAD, ClinVar [67] [69] | Provides allele frequency and clinical interpretation data. | |
| Pathogenicity Predictor | CADD, SIFT, PolyPhen-2 [67] | In silico assessment of variant deleteriousness. | |
| Analysis Databases | Gene Database | OMIM, HGMD [69] | Curated knowledge on gene-disease relationships. |
| Control Cohort | In-house databases / HuaBiao project [67] | Population-matched controls for association studies. |
Both targeted gene panels and WES are powerful tools for uncovering the genetic basis of POI. The decision between them should be guided by the specific clinical and research context. A well-designed 163-gene panel represents an excellent first-tier option for efficient and cost-effective diagnosis when the patient's phenotype strongly suggests a defect in a known POI-associated gene. In contrast, WES is a more comprehensive tool better suited for research discovery, phenotypically complex cases, and when prior panel testing has been non-diagnostic. The integration of data from family trios and the consistent application of ACMG guidelines for variant interpretation are critical for maximizing diagnostic yield, regardless of the platform chosen [67] [69].
Within the context of research on a Next-Generation Sequencing (NGS) panel of 163 Premature Ovarian Insufficiency (POI)-associated genes, establishing clear genotype-phenotype correlations is paramount for translating genetic diagnoses into clinically actionable prognoses. POI, characterized by the loss of ovarian function before age 40, affects approximately 3.5% of the female population and presents with significant heterogeneity in its clinical presentation and residual ovarian function [17]. A genetic diagnosis, achieved in over 29% of cases in large cohorts, is no longer a terminal endpoint but a critical tool for personalized management [5]. This Application Note details how specific genetic findings can inform predictions about a patient's residual ovarian reserve—encompassing both the quantitative follicle pool and qualitative oocyte potential—and outlines standardized protocols for integrating this knowledge into both research and clinical frameworks.
Quantitative Evidence: Genetic Yields and Phenotypic Associations
Large-scale genetic studies have systematically defined the diagnostic yield of POI and begun to link specific genetic etiologies to the prognosis of residual ovarian function. The tables below summarize key quantitative findings.
Table 1: Genetic Diagnostic Yields in POI from Recent Studies
| Cohort / Study | Cohort Size | Diagnostic Yield | Key Genes/Pathways Identified |
|---|---|---|---|
| Large POI Cohort [5] | 375 patients, 70 families | 29.3% | DNA repair genes (e.g., HELQ, C17orf53/HROB, SWI5), BRCA2, FOXL2, BMPR1A/B, novel genes ELAVL2, NLRP11, SPATA33 |
| Idiopathic POI Cohort [3] | 28 patients | 57.1% (Variants of Uncertain Significance included) | FIGLA, TWNK, PMM2, DMC1, MACF1 |
| BPES Patients with POI [74] | 21 patients | 81.0% (with FOXL2 mutations) |
FOXL2 (13 distinct heterozygous variants identified) |
Table 2: Genotype-Phenotype Correlations Informing Ovarian Reserve
| Gene / Variant | Associated Phenotype & Impact on Ovarian Reserve | Key Hormonal / Clinical Biomarkers |
|---|---|---|
FOXL2 mutations [74] |
Type I BPES with POI; Diminished Ovarian Reserve (DOR). Highly heterogeneous reproductive outcomes. | ↑FSH, ↓AMH, ↓AFC, poor response to ovarian stimulation. |
FSHR polymorphisms [75] |
Altered ovarian sensitivity; rs2349415 significantly increases PCOS risk in Punjabi population. Modulates lipid metabolism and hormone levels. |
Modulated LH/FSH levels, associated with dyslipidemia. |
DNA Repair Genes (e.g., HELQ, MSH4) [5] |
POI is often the primary manifestation. Residual reserve can be highly variable. | Standard POI biochemical profile (elevated FSH, low AMH). |
FIGLA pathogenic variant [3] |
Associated with primary amenorrhea and POI, indicating severe ovarian dysfunction from early life. | Greatly elevated FSH (e.g., 58 IU/L), very low AMH (<0.1 ng/mL). |
Experimental Protocols for Genotype-Phenotype Correlation Studies
Protocol 1: Comprehensive Genetic Diagnosis and Analysis
This protocol is adapted from studies that achieved high diagnostic yields using a multi-technique approach [3] [5].
Objective: To identify pathogenic genetic variants in a cohort of POI patients and initiate genotype-phenotype correlation.
Workflow:
Detailed Methodology:
Patient Recruitment and Phenotyping:
- Recruit patients meeting the diagnostic criteria for POI: oligo/amenorrhea for >4 months and elevated FSH >25 IU/L on two occasions >4 weeks apart, before age 40 [17].
- Collect comprehensive clinical data: type of amenorrhea (primary/secondary), age at diagnosis, family history, and baseline hormonal profiles (FSH, LH, E2, AMH).
DNA Extraction:
- Extract genomic DNA from peripheral blood samples using standardized kits (e.g., QIAsymphony DNA midi kits on a QIAsymphony system [3]).
- Quantify DNA using spectrophotometry (e.g., NanoDrop).
Genetic Analysis:
- Array-CGH: Perform oligonucleotide array-CGH (e.g., Agilent SurePrint G3 Human CGH Microarray 4 × 180 K) to identify copy number variations (CNVs). Analyze using software like CytoGenomics and Cartagenia Bench Lab CNV [3].
- Next-Generation Sequencing:
- Library Preparation: Use a custom capture design (e.g., Agilent SureSelect XT-HS) targeting a panel of 163 genes known or suspected in ovarian function [3] [5].
- Sequencing: Sequence on a platform such as Illumina NextSeq 550.
- Bioinformatic Analysis: Utilize pipelines (e.g., Alissa Align&Call) for alignment, variant calling, and annotation. Filter variants against population (gnomAD, DGV) and clinical (ClinVar, HGMD) databases.
Variant Classification and Correlation:
- Classify variants according to American College of Medical Genetics (ACMG) guidelines (Pathogenic, Likely Pathogenic, Variant of Uncertain Significance - VUS) [3].
- Integrate genetic findings with deep phenotyping data to establish correlations, paying special attention to the severity of the phenotype (e.g., primary vs. secondary amenorrhea) in relation to the gene's function.
Protocol 2: Functional Ovarian Reserve Assessment in Genetically-Characterized Patients
Objective: To quantitatively and qualitatively evaluate the residual ovarian reserve in patients with a confirmed genetic diagnosis.
Workflow:
Detailed Methodology:
Biochemical Biomarker Profiling:
- Draw peripheral blood on cycle day 2-4 (or randomly in amenorrhoeic patients).
- Measure serum Anti-Müllerian Hormone (AMH) using an ELISA or automated immunoassay. AMH is a key biomarker of the quantitative ovarian reserve [76] [77].
- Measure basal FSH and estradiol (E2) levels via chemiluminescence immunoassay.
Ultrasonographic Assessment:
- Perform transvaginal ultrasonography in the early follicular phase.
- Count the number of antral follicles (2-10 mm in diameter) in both ovaries to determine the Antral Follicle Count (AFC). For high inter-observer consistency, use 3D automated volume calculation (AVC) technology if available [78].
Ovarian Stimulation Response (if undergoing ART):
- Utilize a standardized controlled ovarian hyperstimulation protocol (e.g., GnRH antagonist protocol with fixed-dose recombinant FSH [78]).
- Record the total gonadotropin (Gn) dosage required and the duration of stimulation.
- Record the number of oocytes retrieved 36 hours post-trigger.
- Calculate the Follicle-to-Oocyte Index (FOI): (Number of oocytes retrieved / Baseline AFC) * 100 [78].
Qualitative Oocyte and Embryo Assessment:
- After retrieval, classify oocyte maturity (Metaphase II).
- Perform fertilization via IVF/ICSI and culture embryos.
- Assess embryo quality morphologically on day 3 (e.g., Veeck criteria) and day 5/6 (e.g., Gardner criteria) [77]. The number of good-quality embryos is a key indicator of oocyte quality.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials and Kits for POI Genetic and Functional Studies
| Item | Function / Application | Example Product / Source |
|---|---|---|
| DNA Extraction Kit | High-quality genomic DNA isolation from whole blood. | QIAsymphony DNA Midi Kits (Qiagen) [3] |
| Array-CGH Platform | Genome-wide detection of copy number variations. | Agilent SurePrint G3 Human CGH Microarray 4 × 180 K [3] |
| NGS Target Enrichment | Custom capture of a 163-gene POI panel for sequencing. | Agilent SureSelect XT-HS Custom Design (Agilent Technologies) [3] |
| NGS Sequencer | High-throughput sequencing of targeted libraries. | Illumina NextSeq 550 System [3] |
| AMH ELISA Kit | Quantitative serum measurement of Anti-Müllerian Hormone. | Kangrun Biotech ELISA Kit [74] |
| Automated 3D Ultrasound | Standardized, operator-independent antral follicle count. | GE Voluson E8 with sonography-based AVC software [78] |
| Recombinant FSH | Standardized ovarian stimulation for response testing. | Gonal-F (Merck KGaA) [78] |
Discussion and Clinical Implications
The integration of a precise genetic diagnosis with a detailed functional assessment of the ovarian reserve allows for a paradigm shift towards personalized medicine in POI.
- Informing Prognosis: Identifying a mutation in a DNA repair gene like
BRCA2versus a transcription factor likeFOXL2carries different prognostic and management implications, including cancer risk [5]. Similarly, the specific type ofFOXL2mutation can help predict the severity of ovarian dysfunction, though significant heterogeneity exists [74]. - Guiding Therapeutic Interventions: A confirmed genetic diagnosis can direct patients towards the most appropriate fertility interventions. For example, patients with a low quantitative but potentially preserved qualitative reserve might be candidates for innovative techniques like in vitro activation (IVA) [5].
- Predictive Modeling: Machine learning models are now being developed that integrate genetic data with clinical features (age, AMH, AFC) and serum biomarkers (e.g., GDF9, BMP15, oxidative stress markers) to more accurately predict both the quantity and quality of the ovarian reserve, outperforming single markers like AMH alone [77]. The genetic profile is a key feature in these models.
In conclusion, the systematic application of the protocols and correlations described herein empowers researchers and clinicians to move beyond a simple diagnosis of POI. It enables the stratification of patients based on their underlying genetic etiology and provides a data-driven estimate of their residual ovarian potential, thereby facilitating improved counseling, personalized therapeutic strategies, and long-term health management.
Premature Ovarian Insufficiency (POI) is a complex clinical disorder characterized by the loss of ovarian function before age 40, affecting approximately 1-3.7% of women and presenting significant fertility and health challenges [3] [79]. Despite its clinical importance, the etiology of approximately 70% of POI cases remains unexplained, creating a substantial barrier to effective therapeutic development [3]. Advances in genetic research, particularly through next-generation sequencing (NGS) panels targeting POI-associated genes, have begun to illuminate the molecular basis of this condition. The integration of these genetic findings with systematic drug target identification methodologies provides a powerful framework for translating gene discovery into viable therapeutic strategies. This Application Note outlines established protocols for identifying and validating druggable pathways and compounds derived from NGS-based genetic research, offering a structured approach for researchers and drug development professionals working within the context of a 163-gene POI panel.
Genetic Screening and Target Identification Protocols
NGS Panel Screening for POI
Objective: To identify pathogenic genetic variants in idiopathic POI patients using a targeted NGS approach.
Materials:
- Patient DNA samples from peripheral blood (28 idiopathic POI patients recommended)
- Custom NGS capture design for 163 genes associated with ovarian function
- SureSelect XT-HS reagents (Agilent Technologies)
- NextSeq 550 sequencing system (Illumina)
- Alissa Align&Call v1.1 and Alissa Interpret v5.3 software
Procedure:
- Patient Selection: Include women meeting diagnostic criteria for POI (primary or secondary amenorrhea >4 months before age 40 with FSH >25 IU/L). Exclude patients with karyotype abnormalities, FMR1 premutations, or known autoimmune/iatrogenic causes [3].
- DNA Extraction: Isolate DNA from peripheral blood using QIAsymphony DNA midi kits on a QIAsymphony system.
- Library Preparation & Sequencing: Utilize SureSelect XT-HS reagents with custom capture design following manufacturer's protocols. Sequence on NextSeq 550 system.
- Bioinformatic Analysis:
- Perform sequence alignment and variant calling using Alissa Align&Call.
- Annotate variants and filter against population databases (gnomAD, DGV).
- Cross-reference with variant databases (ClinVar, HGMD) and literature.
- Variant Classification: Classify variants according to ACMG guidelines (Pathogenic, Likely Pathogenic, VUS, Likely Benign, Benign).
Expected Outcomes: A recent study implementing this protocol identified causal genetic anomalies in 57.1% (16/28) of idiopathic POI patients, including single nucleotide variations (SNVs), indels, and copy number variations (CNVs) [3].
Integration of Array-CGH for Comprehensive Genetic Analysis
Objective: To detect copy number variations that may be missed by NGS alone.
Materials:
- SurePrint G3 Human CGH Microarray 4 × 180 K (Agilent Technologies)
- CytoGenomics software v5.0
- Cartagenia Bench Lab CNV software v5.1
Procedure:
- Hybridization: Perform oligonucleotide array-CGH according to manufacturer specifications.
- CNV Detection: Use Feature Extraction and CytoGenomics software with standard settings (minimum 60 kb detection threshold).
- Annotation & Interpretation: Analyze identified CNVs using Cartagenia Bench Lab CNV, referencing DECIPHER and ClinGen databases.
Expected Outcomes: Combined NGS and array-CGH analysis increases diagnostic yield, with one study identifying CNVs in 3.6% (1/28) of POI patients [3].
Quantitative Analysis of Genetic Screening Data
Table 1: Genetic Findings from Combined NGS and Array-CGH Analysis in POI Patients
| Analysis Method | Patients with Causal Variants | Variant of Uncertain Significance (VUS) | Key Example Genes Identified |
|---|---|---|---|
| Array-CGH (CNVs) | 3.6% (1/28) | 7.1% (2/28) | 15q25.2 deletion |
| NGS (SNVs/Indels) | 28.6% (8/28) | 25% (7/28) | FIGLA, TWNK, PMM2 |
| Combined Approach | 57.1% (16/28) | 25% (7/28) | Multiple |
Table 2: Clinical Characteristics of POI Cohort for Genetic Screening
| Parameter | Value | Note |
|---|---|---|
| Total Patients | 28 | Idiopathic POI |
| Primary Amenorrhea | 14.3% (4/28) | - |
| Secondary Amenorrhea | 85.7% (24/28) | - |
| Average Age at Diagnosis | 27.7 years | - |
| Family History of POI | 39.3% (11/28) | Suggests genetic component |
Druggable Target Prioritization Framework
Mendelian Randomization and Colocalization Analysis
Objective: To establish causal relationships between gene expression and POI risk using genetic instruments.
Materials:
- Cis-eQTL data from GTEx V8 (ovary and whole blood) and eQTLGen consortium
- POI GWAS data from FinnGen study (599 cases, 241,998 controls)
- SMR software (version 1.3.1)
- coloc R package
Procedure:
- Data Acquisition:
- Download cis-eQTL data for 431 genes with index cis-eQTL signals (P~eQTL~ < 5 × 10^-8) from GTEx portal and eQTLGen.
- Obtain POI GWAS summary statistics from FinnGen R11 dataset.
- Summary-based MR (SMR) Analysis:
- Perform SMR to test associations between gene expression and POI risk.
- Conduct HEIDI test to detect pleiotropy (P~HEIDI~ < 0.05 indicates exclusion).
- Colocalization Analysis:
- Use Bayesian approach to calculate posterior probabilities for five hypotheses (PP.H0-PP.H4).
- Apply priors: p1 = 1 × 10^-4, p2 = 1 × 10^-4, p12 = 1 × 10^-5.
- Consider strong evidence when PP.H3 + PP.H4 ≥ 0.8.
Expected Outcomes: This protocol identified FANCE and RAB2A as promising therapeutic targets with strong colocalization evidence (PP.H3 + PP.H4 ≥ 0.8) [79].
Druggability Assessment Protocol
Objective: To evaluate the potential of candidate genes as drug targets.
Materials:
- Online Mendelian Inheritance in Man (OMIM) database
- DrugBank database
- Drug-Gene Interaction database (DGIdb)
- Therapeutic Target Database (TTD)
Procedure:
- Database Query: Interrogate each database for known drug interactions, clinical trial status, and mechanistic information.
- Assessment Criteria:
- Category 1: Approved for marketing or in clinical trials
- Category 2: In preclinical development
- Category 3: Considered druggable based on protein family/function
- Pathway Analysis: Map gene function to biological pathways (e.g., DNA repair, autophagy) to identify broader therapeutic opportunities.
Expected Outcomes: Recent analysis identified FANCE (involved in DNA repair) and RAB2A (regulates autophagy) as druggable candidates for POI, though not yet targeted in clinical practice [79].
Inflammation-Related Protein Causal Analysis
Objective: To identify inflammation-related proteins with causal effects on POI risk using Mendelian randomization.
Materials:
- Genetic instruments for 91 inflammation-related proteins from Olink Target Inflammation panel (14,824 participants)
- POI GWAS data from FinnGen (424 cases, 118,796 controls)
- TwoSampleMR R package
Procedure:
- Instrument Selection: Identify SNPs associated with inflammation-related proteins at genome-wide significance (P < 5 × 10^-8).
- LD Clustering: Apply linkage disequilibrium clustering (R^2^ < 0.001, distance = 10,000 kb).
- MR Analysis:
- Perform inverse-variance weighted (IVW) method as primary analysis.
- Conduct sensitivity analyses (MR-Egger, weighted median, MR-PRESSO).
- Apply Bonferroni correction (P < 1 × 10^-4 for significance).
- Experimental Validation:
- Treat KGN human granulosa-like tumor cells with 1 mg/mL cyclophosphamide for 48h to model POI.
- Validate protein expression changes via Western blot.
Expected Outcomes: This approach identified CXCL10 and CX3CL1 as protective against POI, while IL-18R1, IL-18, MCP-1, and CCL28 increase POI risk [80].
Table 3: Druggable Target Assessment for POI-Associated Genes
| Gene | MR Evidence | Colocalization Evidence | Biological Function | Druggability Assessment |
|---|---|---|---|---|
| FANCE | Significant (P < 0.05) | Strong (PP.H3+PP.H4 ≥ 0.8) | DNA repair, Fanconi anemia pathway | Preclinical candidate |
| RAB2A | Significant (P < 0.05) | Strong (PP.H3+PP.H4 ≥ 0.8) | Autophagy regulation, vesicular trafficking | Novel target |
| HM13 | Significant (P < 0.05) | Weak | Signal peptide peptidase | Limited evidence |
| MLLT10 | Significant (P < 0.05) | Weak | Chromatin modification, transcription | Limited evidence |
Table 4: Inflammation-Related Protein Targets for POI
| Protein | Causal Relationship with POI | Potential Therapeutic Approach | Experimental Validation in POI Model |
|---|---|---|---|
| MCP-1/CCL2 | Risk factor | Inhibition | Significantly changed in POI model |
| TGFB1 | Risk factor | Inhibition | Significantly changed in POI model |
| ARTN | Risk factor | Inhibition | Significantly changed in POI model |
| LIFR | Risk factor | Inhibition | Significantly changed in POI model |
| CXCL10 | Protective | Augmentation | Not tested |
Experimental Validation Workflows
CRISPR-Drug Perturbational Screening Protocol
Objective: To identify druggable gene knockouts that sensitize cells to existing chemotherapeutic agents.
Materials:
- Targeted CRISPR library (655 druggable genes, 6 gRNAs/gene)
- Cas9-expressing cell lines (10 neuroblastoma, 8 non-neuroblastoma recommended)
- Chemotherapeutic agents (doxorubicin, topotecan, cisplatin, etc.)
- Next-generation sequencing platform
- MAGeCK software for analysis
Procedure:
- Library Design: Design gRNAs targeting druggable genes based on established druggable genomes [81].
- Cell Line Selection: Include diverse cell types covering disease-relevant mutations (e.g., TP53) and resistance phenotypes.
- Screening Setup:
- Transduce cells with CRISPR library at low MOI to ensure single integrations.
- Treat with IC~20~-IC~30~ drug concentrations for 14-21 days.
- Include vehicle-treated controls.
- Sequencing & Analysis:
- Harvest genomic DNA and amplify gRNA regions for sequencing.
- Use MAGeCK to calculate robust ranking aggregation (RRA) scores.
- Identify sensitizing knockouts (negative RRA) and resistance mechanisms (positive RRA).
Expected Outcomes: This approach generated 94,320 unique combination-cell line perturbations, successfully identifying PRKDC inhibition as sensitizing neuroblastoma cells to doxorubicin both in vitro and in vivo [81].
In Vitro Validation in Ovarian Cell Models
Objective: To validate candidate drug targets in ovarian-relevant cellular contexts.
Materials:
- KGN human granulosa-like tumor cell line
- Cyclophosphamide (1 mg/mL) for POI modeling
- Antibodies for target proteins (MCP-1, TGF-β1, ARTN, LIF-R)
- RPMI 1640 culture medium
- Western blot and RT-PCR equipment
Procedure:
- Cell Culture: Maintain KGN cells in RPMI 1640 medium at 37°C with 5% CO~2~.
- POI Model Induction: Treat cells with 1 mg/mL cyclophosphamide for 48 hours.
- Protein Analysis:
- Extract total protein using RIPA buffer.
- Separate proteins by SDS-PAGE and transfer to PVDF membranes.
- Probe with primary antibodies (1:500-1:50,000 dilution).
- Detect with HRP-conjugated secondary antibodies (1:10,000).
- Gene Expression Analysis:
- Extract RNA using TRIzol method.
- Perform reverse transcription and quantitative PCR.
- Analyze expression changes relative to housekeeping genes.
Expected Outcomes: This protocol confirmed significant changes in MCP-1/CCL2, TGFB1, ARTN, and LIFR in the POI model, converging on the oncostatin M signaling pathway [80].
Therapeutic Development Strategies
Drug Repurposing and Combination Therapy Approach
Objective: To identify existing drugs that can be repurposed for POI treatment based on genetic evidence.
Materials:
- DGIdb database for drug-gene interactions
- Clinical compound libraries
- In vitro ovarian follicle culture system
Procedure:
- Target-Drug Mapping:
- Query DGIdb for known inhibitors/activators of prioritized targets.
- Filter for compounds with FDA approval or late-stage clinical development.
- Combination Screening:
- Test candidate drugs in combination with standard agents in relevant cellular models.
- Assess synergy using Chou-Talalay method.
- Functional Assessment:
- Utilize ovarian follicle culture systems to evaluate effects on follicle activation and growth.
- Measure markers of folliculogenesis (AMH, FSH receptor expression).
Expected Outcomes: Gene-drug analysis identified CCL2 and TGFB1 as potential therapeutic targets, with genistein and melatonin prioritized as potential drugs for POI treatment [80].
The Scientist's Toolkit: Research Reagent Solutions
Table 5: Essential Research Reagents for POI Drug Target Identification
| Reagent/Category | Specific Examples | Function in POI Research |
|---|---|---|
| NGS Panels | Custom 163-gene panel (POI-associated) | Comprehensive screening of known POI genes |
| CRISPR Libraries | Targeted druggable genome library (655 genes) | High-throughput identification of gene-drug interactions |
| Cell Models | KGN human granulosa-like tumor cell line | In vitro validation of targets in ovarian context |
| POI Induction Agent | Cyclophosphamide (1 mg/mL) | Chemical induction of POI phenotype in cellular models |
| Bioinformatic Tools | SMR, MAGeCK, TwoSampleMR, coloc | Statistical analysis of genetic and multi-omics data |
| Validation Antibodies | Anti-MCP-1, Anti-TGF-β1, Anti-LIF-R | Protein-level confirmation of target expression |
Visualization of Workflows and Pathways
Genetic Screening to Drug Target Identification Workflow
Inflammation Pathway in POI Pathogenesis
CRISPR-Drug Screening Methodology
Application Note
This application note details a framework for validating novel candidate genes implicated in Premature Ovarian Insufficiency (POI), specifically within the context of research utilizing a next-generation sequencing (NGS) panel of 163 POI-associated genes. The validation of novel genes identified through high-throughput sequencing is a critical step in translating genetic findings into clinically actionable insights and understanding underlying biological mechanisms.
Recent large-scale cohort studies have demonstrated the efficacy of a multi-modal genetic approach for diagnosing idiopathic POI. A 2025 study involving 28 patients with idiopathic POI, which combined array-CGH and a custom NGS panel of 163 genes, successfully identified a genetic anomaly in 57.1% (16 of 28) of patients [3]. This high diagnostic yield underscores the power of comprehensive genetic testing in a disorder where a significant proportion of cases remain unexplained. The study employed a rigorous variant classification system according to the American College of Medical Genetics (ACMG) standards, identifying causal single nucleotide variations (SNVs) or indels in 28.6% (8 of 28) of patients and a causal copy number variation (CNV) in at least one patient [3]. The remaining seven patients carried variants of uncertain significance (VUS), highlighting the ongoing need for functional validation and data sharing to reclassify these findings [3].
The success of such studies is highly dependent on the quality of the biospecimens and the analytical sensitivity of the NGS platform. For instance, in the context of cancer genomics, a prospective multicenter trial (cPANEL) demonstrated that cytology specimens preserved in a nucleic acid stabilizer could achieve a 98.4% success rate for gene panel analysis, with a high positive concordance rate of 97.3% compared to other diagnostic methods [82]. This highlights the importance of optimized sample handling and sensitive panel design, principles that are directly transferable to POI research. The analytical performance of a panel, including its limit of detection (LOD) for specific variant types, is a cornerstone of reliable gene validation [82].
Table 1: Key Performance Metrics from Genetic Studies of Idiopathic POI
| Study Parameter | Result | Implication for Validation |
|---|---|---|
| Patients with a Genetic Anomaly | 16/28 (57.1%) [3] | Confirms high genetic burden and justifies NGS panel use. |
| Causal SNVs/Indels Identified | 8/28 (28.6%) [3] | Demonstrates the panel's ability to detect monogenic causes. |
| Causal CNV Identified | 1/28 [3] | Validates the necessity of a combined SNV and CNV approach. |
| Variants of Uncertain Significance (VUS) | 7/28 [3] | Highlights candidates for future validation studies. |
| Example Pathogenic Gene | FIGLA [3] | Provides a positive control for panel performance. |
Experimental Protocols
Patient Cohort and Sample Preparation
The foundation of any robust genetic validation study is a well-characterized patient cohort and high-quality nucleic acid extraction.
- Patient Cohort Definition: The study should include patients with a confirmed diagnosis of idiopathic POI, characterized by primary or secondary amenorrhea of more than 4 months before the age of 40 years and elevated follicle-stimulating hormone (FSH) levels (>25 IU/L) [3]. A critical step is the exclusion of patients with known karyotype abnormalities, FMR1 gene premutations, or identifiable iatrogenic/autoimmune causes to ensure the cohort is truly idiopathic and enriched for novel genetic causes [3].
- Sample Collection and DNA Extraction: Peripheral blood samples are collected from participants after obtaining informed consent. DNA is extracted from leukocytes using standardized commercial kits, such as those from Qiagen (e.g., QIAsymphony DNA midi kits on a QIAsymphony system), to ensure purity and yield suitable for NGS library preparation [3]. The quality and quantity of DNA should be assessed using fluorometry (e.g., Qubit) and spectrophotometry (e.g., NanoDrop) [82].
Multi-Modal Genetic Analysis
This protocol employs a combination of techniques to capture a wide spectrum of genetic variation, from single nucleotides to large chromosomal rearrangements.
- Array-CGH for CNV Detection: Oligonucleotide array-CGH is performed using platforms like the SurePrint G3 Human CGH Microarray 4 × 180 K (Agilent Technologies). This technique can detect CNVs as small as 60 kb across the genome. Bioinformatic analysis is conducted using dedicated software (e.g., CytoGenomics, Agilent Technologies), and identified CNVs are interpreted with database resources such as DECIPHER and ClinGen [3].
- Targeted NGS Panel for SNV and Indel Detection:
- Library Preparation: A custom SureSelect XT-HS capture design (Agilent Technologies) targeting the 163 POI-associated genes is used. Library preparation is performed according to the manufacturer's recommendations on a system such as the Magnis (Agilent Technologies) [3].
- Sequencing: The prepared libraries are sequenced on a high-throughput platform like the Illumina NextSeq 550 system [3].
- Bioinformatic Analysis: Sequencing reads are aligned to a reference genome (e.g., GRCh37/hg19), and variants are called using software such as Alissa Align&Call and interpreted with Alissa Interpret (Agilent Technologies) [3].
Variant Filtering and Classification
The analytical workflow for identifying and prioritizing variants from raw sequencing data is crucial for distinguishing true pathogenic variants from benign polymorphisms.
Variant Interpretation Workflow
- Variant Annotation and Filtering: Called variants are annotated against population databases (e.g., gnomAD), variation databases (e.g., ClinVar, HGMD), and the literature [3]. Initial filtering removes common polymorphisms (e.g., population allele frequency >1%) and focuses on rare, protein-altering variants (non-synonymous, splice-site, indels) within the 163-gene panel.
- ACMG Classification: Each remaining variant is classified according to ACMG guidelines into one of five categories: benign, likely benign, variant of uncertain significance (VUS), likely pathogenic, or pathogenic [3]. This standardized classification is essential for clinical interpretation and for selecting candidates for further functional validation. The identification of novel variants in known POI genes, as seen in studies on epilepsy panels (e.g., novel variants in DEPDC5, SCN1A), reinforces the value of this approach in expanding the mutational spectrum of disease-associated genes [83].
Table 2: Research Reagent Solutions for NGS Panel Validation
| Reagent/Kit | Function in Protocol | Example Product |
|---|---|---|
| Nucleic Acid Extraction Kit | Purifies high-quality DNA from whole blood. | QIAsymphony DNA Midi Kits (Qiagen) [3] |
| Array-CGH Platform | Detects genome-wide copy number variations (CNVs). | SurePrint G3 Human CGH Microarray 4x180K (Agilent Technologies) [3] |
| Targeted Capture Kit | Enriches for the 163 POI-associated genes prior to sequencing. | SureSelect XT-HS Custom Design (Agilent Technologies) [3] |
| NGS Sequencing System | Performs high-throughput sequencing of prepared libraries. | NextSeq 550 System (Illumina) [3] |
| Variant Interpretation Software | Aids in alignment, variant calling, and annotation. | Alissa Align&Call & Alissa Interpret (Agilent Technologies) [3] |
Data Integration and Pathway Analysis
Following the identification of high-confidence genetic variants, the final step involves integrating these findings into a biological context to elucidate disrupted molecular pathways and prioritize candidates for functional studies.
The candidate genes identified through the NGS panel should be analyzed for enrichment in specific biological pathways critical for ovarian function. The candidate gene approach is strengthened when genes can be logically linked to the disease's pathophysiology, such as pathways involved in oogenesis, folliculogenesis, meiosis, and DNA repair [3]. For example, a gene like FIGLA is a established transcription factor in ovarian development, and the finding of a pathogenic variant (e.g., a homozygous duplication causing a frameshift) provides a direct molecular diagnosis [3]. Constructing a signaling pathway map that incorporates both known genes (e.g., NOBOX, BMP15) and novel candidates from the study helps generate testable hypotheses about disease mechanisms.
POI Candidate Gene Pathway Integration
This integrated approach, combining rigorous cohort design, multi-modal genetic testing, and careful bioinformatic and pathway analysis, provides a powerful and validated protocol for uncovering the genetic architecture of idiopathic POI. It directly facilitates the translation of genetic discoveries into improved diagnostics, genetic counseling for families, and targets for future therapeutic interventions [3].
Conclusion
The deployment of a comprehensive 163-gene NGS panel represents a paradigm shift in POI research, moving a substantial number of cases from 'idiopathic' to 'explained' and achieving a genetic diagnosis in over 57% of patients when combined with array-CGH. This genetic dissection reveals a complex landscape dominated by defects in meiotic and DNA repair pathways, with a significant subset of findings carrying implications for lifelong health beyond fertility, such as cancer predisposition. For researchers, these panels are powerful tools for gene discovery and functional validation. For drug development, they unveil novel, genetically-defined therapeutic targets, such as those in inflammatory pathways, and enable the repurposing of existing agents. The future of POI management lies in this genetically-informed, personalized approach, which promises not only to refine diagnostic and prognostic accuracy but also to open new avenues for interventions aimed at preserving fertility and delaying ovarian aging.
References
- 1. Changing Etiological Spectrum of Premature Ovarian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12248835/]
- 2. Premature ovarian insufficiency — Knowledge Hub [https://www.genomicseducation.hee.nhs.uk/genotes/knowledge-hub/premature-ovarian-insufficiency/]
- 3. Genetics Investigation of Idiopathic Premature Ovarian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11942377/]
- 4. Next-generation sequencing of 500 POI patients identified ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9930292/]
- 5. Genetic landscape of a large cohort of Primary Ovarian ... [https://www.sciencedirect.com/science/article/pii/S2352396422004285]
- 6. DNA double-strand break genetic variants in patients with ... [https://ovarianresearch.biomedcentral.com/articles/10.1186/s13048-023-01221-2]
- 7. Targeted Gene Sequencing Panels [https://emea.illumina.com/techniques/sequencing/dna-sequencing/targeted-resequencing/targeted-panels.html]
- 8. Frontiers | Bioinformatics and Immune Infiltration Analyses Reveal the... [https://www.frontiersin.org/journals/cardiovascular-medicine/articles/10.3389/fcvm.2021.696321/full]
- 9. Identification of potential key , pathways and circulating markers... genes [https://pubmed.ncbi.nlm.nih.gov/32589050/]
- 10. Multi-omics strategies for biomarker discovery and ... [https://link.springer.com/article/10.1186/s43556-025-00340-0]
- 11. Premature ovarian insufficiency: clinical orientations for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7722400/]
- 12. Premature Ovarian Failure Panel [https://www.blueprintgenetics.com/tests/panels/endocrinology/premature-ovarian-failure-panel/]
- 13. Genetics, Autosomal Dominant - StatPearls - NCBI Bookshelf [https://www.ncbi.nlm.nih.gov/books/NBK557512/]
- 14. Comprehensive NGS Panel Validation for the Identification ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8145002/]
- 15. A targeted next-generation sequencing panel for ... [https://www.nature.com/articles/s41598-025-08039-6]
- 16. Genetics of Primary Ovarian Insufficiency in the Next ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7033037/]
- 17. Evidence-based guideline: Premature Ovarian Insufficiency [https://www.asrm.org/practice-guidance/practice-committee-documents/evidence-based-guideline-premature-ovarian-insufficiency--2024/]
- 18. Noninvasive markers for warning premature ovarian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12150474/]
- 19. Next-generation sequencing of 500 POI patients identified ... [https://ovarianresearch.biomedcentral.com/articles/10.1186/s13048-023-01104-6]
- 20. Targeted Next-Generation Sequencing Indicates a ... [https://www.frontiersin.org/journals/endocrinology/articles/10.3389/fendo.2021.664645/full]
- 21. Identifying susceptibility genes for primary ovarian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8494118/]
- 22. Identification of variants in pleiotropic genes causing “ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6117257/]
- 23. Polygenic risk scores: from research tools to clinical instruments [https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-020-00742-5]
- 24. Primary ovarian insufficiency: update on clinical and ... [https://www.frontiersin.org/journals/endocrinology/articles/10.3389/fendo.2024.1464803/full]
- 25. Guidelines for Validation of Next-Generation Sequencing– ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6941185/]
- 26. Next-Generation Sequencing (NGS) | Explore the technology [https://www.illumina.com/science/technology/next-generation-sequencing.html]
- 27. Next-Generation Sequencing Technology: Current Trends ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10376292/]
- 28. Sequencing Data Analysis | NGS software to help you ... [https://www.illumina.com/informatics/sequencing-data-analysis.html]
- 29. Array CGH-based detection of CNV regions and their potential ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-019-5552-1]
- 30. Comparison of CNV analysis methods: Array CGH vs NGS [https://3billion.io/blog/comparison-of-cnv-analysis-methods-array-cgh-vs-ngs]
- 31. Next generation sequencing and array-based comparative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5495984/]
- 32. Frontiers | Experiences from dual genome next-generation sequencing... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1488956/full]
- 33. Screening of premature ovarian insufficiency associated in... genes [https://pubmed.ncbi.nlm.nih.gov/38649916/]
- 34. Whole genome sequencing for copy number variant ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11625463/]
- 35. Improving CNV Detection Performance in Microarray Data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10871075/]
- 36. Comparative study of aCGH and Next Generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4510565/]
- 37. Copy number variation detection using next generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4021345/]
- 38. Genetic variants in HELB contribute to premature ovarian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12643492/]
- 39. A Bioinformatics Toolkit for Next-Generation Sequencing in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10741970/]
- 40. Variant Calling Pipeline: FastQ to Annotated SNPs in Hours [https://gencore.bio.nyu.edu/variant-calling-pipeline/]
- 41. Bioinformatics Pipeline: DNA-Seq Analysis: Whole Exome and ... [https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/DNA_Seq_Variant_Calling_Pipeline/]
- 42. Part 1: Per-sample variant calling [https://training.nextflow.io/2.4.0/nf4_science/genomics/01_per_sample_variant_calling/]
- 43. Best practices for variant calling in clinical sequencing [https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-020-00791-w]
- 44. MGA loss-of-function variants cause premature ovarian ... [https://www.jci.org/articles/view/183758]
- 45. and biallelic variant effects on disease at biobank scale [https://www.nature.com/articles/s41586-022-05420-7]
- 46. Whole-exome sequencing in patients with premature ovarian ... [https://ovarianresearch.biomedcentral.com/articles/10.1186/s13048-020-00716-6]
- 47. Personalised genomic strategies improve diagnostic yield ... [https://www.nature.com/articles/s41433-025-03981-1]
- 48. Monoallelic and Biallelic Variants in EMC1 Identified in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4800043/]
- 49. Variants of uncertain significance: At the crux of diagnostic ... [https://www.sciencedirect.com/science/article/abs/pii/S0378111925003762]
- 50. Landscape of pathogenic mutations in premature ovarian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9941050/]
- 51. Increasing pathogenic germline variant diagnosis rates in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12374290/]
- 52. Genome-wide prediction of disease variant effects with a ... [https://www.nature.com/articles/s41588-023-01465-0]
- 53. Integrating next-generation sequencing and artificial ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2025.1568205/full]
- 54. A Comparative Analysis of Two Cohorts from a Single Center [https://www.mdpi.com/2075-4418/15/13/1724]
- 55. The landscape of genetic variations in non-syndromic ... [https://www.frontiersin.org/journals/endocrinology/articles/10.3389/fendo.2023.1289333/full]
- 56. Secondary Amenorrhea - StatPearls - NCBI Bookshelf [https://www.ncbi.nlm.nih.gov/books/NBK431055/]
- 57. Identification of genetic variants in patients with primary ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12199654/]
- 58. Genome-wide insights of shared genetic architecture ... [https://www.sciencedirect.com/org/science/article/pii/S2049361425001613]
- 59. Guidance on Incidental Findings [https://bsgm.org.uk/healthcare-professionals/guidance-on-incidental-findings/]
- 60. Incidental findings from cancer next generation sequencing ... [https://www.nature.com/articles/s41525-021-00224-6]
- 61. ACMG SF v3.3 list for reporting of secondary findings in ... [https://pubmed.ncbi.nlm.nih.gov/40568962/]
- 62. Knowing When to Respond to Incidental Findings [https://www.rsna.org/news/2025/august/when-to-respond-to-incidental-findings]
- 63. Genetic Modifiers and Oligogenic Inheritance - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4448705/]
- 64. From Peas to Disease: Modifier Genes, Network Resilience ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5544383/]
- 65. Next-generation sequencing and its clinical application [https://pmc.ncbi.nlm.nih.gov/articles/PMC6528456/]
- 66. The genetic modifier approach: identifying the right target [https://www.drugtargetreview.com/article/110341/the-genetic-modifier-approach-identifying-the-right-target-for-rare-diseases/]
- 67. Landscape of pathogenic mutations in premature ovarian ... [https://www.nature.com/articles/s41591-022-02194-3]
- 68. Targeted gene panel provides advantages over whole-exome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10847719/]
- 69. Enhanced utility of family-centered diagnostic exome ... [https://www.nature.com/articles/gim2014154]
- 70. A meta-analysis of diagnostic yield and clinical utility ... [https://www.sciencedirect.com/science/article/pii/S1098360025000450]
- 71. Next generation sequencing panel as an effective approach to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11249508/]
- 72. Whole-exome sequencing reveals new potential genes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8995228/]
- 73. Novel variants associated with premature ovarian ... [https://www.frontiersin.org/journals/endocrinology/articles/10.3389/fendo.2025.1687148/full]
- 74. Ovarian Reserve and ART Outcomes in Blepharophimosis ... [https://www.frontiersin.org/journals/endocrinology/articles/10.3389/fendo.2022.829153/full]
- 75. Exploring genotype-phenotype correlation of FSHR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12548221/]
- 76. Anti-Müllerian hormone as a biomarker of ovarian function ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12477649/]
- 77. Assessment and prediction models for the quantitative and ... [https://ovarianresearch.biomedcentral.com/articles/10.1186/s13048-025-01732-0]
- 78. Impact of gonadotropin genetic profile and ovarian reserve on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12411160/]
- 79. Identifying therapeutic targets for primary ovarian insufficiency ... [https://ovarianresearch.biomedcentral.com/articles/10.1186/s13048-024-01524-y]
- 80. Genetic Association and Drug Target Exploration Between ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12517199/]
- 81. A CRISPR-drug perturbational map for identifying ... [https://www.nature.com/articles/s41467-023-43134-0]
- 82. Prospective multicenter validation of a next-generation ... [https://bmccancer.biomedcentral.com/articles/10.1186/s12885-025-14770-0]
- 83. Validation of targeted next-generation sequencing panels ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10849089/]