Mendelian Randomization for POI: Unraveling Causal Genes and Informing Therapeutic Development
Primary Ovarian Insufficiency (POI) is a significant cause of female infertility, affecting 1-3% of women, yet its etiology remains largely elusive.
Mendelian Randomization for POI: Unraveling Causal Genes and Informing Therapeutic Development
Abstract
Primary Ovarian Insufficiency (POI) is a significant cause of female infertility, affecting 1-3% of women, yet its etiology remains largely elusive. This article synthesizes the latest research applying Mendelian Randomization (MR) to elucidate the causal genetic architecture of POI. We explore foundational discoveries of pathogenic variants in genes governing meiosis, folliculogenesis, and immune function, and detail the methodological application of MR integrated with expression quantitative trait loci (eQTL) data for causal inference and drug target prioritization. The review critically addresses common analytical pitfalls in drug-target MR and provides optimization strategies to ensure robust findings. Finally, we examine how MR findings are validated through colocalization analysis and comparative studies, positioning MR as a powerful tool for de-risking drug development by identifying genetically validated therapeutic targets for POI, such as FANCE and RAB2A.
The Genetic Landscape of POI: Establishing a Causal Foundation
POI Clinical Definition and the Challenge of Idiopathic Cases
Clinical and Epidemiological Framework of POI
Premature ovarian insufficiency (POI) is a significant clinical disorder characterized by the loss of ovarian function before the age of 40. The condition is diagnosed based on the following core criteria: oligomenorrhea or amenorrhea for at least 4 months, and elevated follicle-stimulating hormone (FSH) levels >25 IU/L on two occasions more than 4 weeks apart [1] [2]. This definition aligns with guidelines established by the European Society of Human Reproduction and Embryology (ESHRE) [1].
POI affects women's health comprehensively, leading to short-term symptoms including infertility, menstrual disturbances, vasomotor symptoms (hot flashes, night sweats), mood changes, vaginal dryness, and decreased quality of life [1] [3]. Long-term health consequences include increased risks of osteoporosis, cardiovascular disease, cognitive decline, and premature mortality due to prolonged hypoestrogenism [1] [4].
The global prevalence of POI is approximately 3.7%, though estimates vary across different populations and studies, ranging from 1% to 3.7% of women under 40 [1] [3] [2]. Epidemiological data reveal distinct patterns across age groups and ethnicities. The incidence declines exponentially with decreasing age, affecting approximately 1:100 women aged 35-40, 1:1,000 women aged 25-30, and 1:10,000 women aged 18-25 [4]. Recent studies suggest the incidence among younger women may be increasing [4].
Table 1: Global Epidemiology of Premature Ovarian Insufficiency
| Population | Prevalence | Key Epidemiological Notes |
|---|---|---|
| Global Estimate | 3.7% | Based on recent meta-analyses [1] [2] |
| Women under 40 | 1% | Broader historical estimate [3] |
| By Age | Varies exponentially | 1:100 (35-40 yrs); 1:1,000 (25-30 yrs); 1:10,000 (18-25 yrs) [4] |
| Ethnic Variations | Higher in Hispanic, African American | Lower prevalence in Japanese, Chinese populations [4] |
| Regional Examples | 1.9% (Sweden), 3.5% (Iran) | Demonstrates geographical variability [4] |
The Complex Etiological Spectrum and Idiopathic Challenge
POI has a multifactorial etiological background encompassing genetic abnormalities, autoimmune disorders, and induced damage to the ovarian follicular reserve. The distribution of causes has evolved significantly over recent decades, with a notable decrease in idiopathic cases due to improved diagnostic capabilities [1].
Table 2: Etiological Distribution of POI: Historical vs. Contemporary Cohorts
| Etiology | Historical Cohort (1978-2003) | Contemporary Cohort (2017-2024) | Statistical Significance |
|---|---|---|---|
| Genetic | 11.6% | 9.9% | Not Significant |
| Autoimmune | 8.7% | 18.9% | Significant (p<0.05) |
| Iatrogenic | 7.6% | 34.2% | Significant (p<0.05) |
| Idiopathic | 72.1% | 36.9% | Significant (p<0.05) |
The most substantial change in the etiological landscape is the more than fourfold rise in identifiable iatrogenic cases and a twofold increase in the autoimmune group, resulting in a halving of idiopathic POI [1]. Despite these diagnostic advances, a substantial proportion of cases (approximately 23.5-36.9%) remain classified as idiopathic [1] [2], underscoring the ongoing challenge in POI research and clinical management.
Established Etiological Categories
Genetic Causes: Chromosomal abnormalities, particularly X-chromosome anomalies such as Turner syndrome, account for approximately 12-13% of POI cases [1]. The fragile X premutation (FMR1 gene) affects 20-30% of carriers, with risk influenced by CGG repeat size [1]. Research has identified mutations in more than 75 genes associated with POI, primarily involved in meiosis, DNA repair, and ovarian development [1] [2]. Whole-exome sequencing studies have demonstrated that pathogenic variants in known POI-causative genes account for approximately 18.7% of cases [2], with a higher diagnostic yield (25.8%) in primary amenorrhea compared to secondary amenorrhea (17.8%) [2].
Autoimmune Causes: Autoimmune mechanisms contribute to approximately 4-30% of spontaneous POI cases [1]. Common associated conditions include Hashimoto's thyroiditis, Addison's disease, Graves' disease, type 1 diabetes mellitus, rheumatoid arthritis, and systemic lupus erythematosus [1] [3]. Hashimoto's thyroiditis confers an 89% higher risk of amenorrhea and a 2.4-fold increased risk of infertility due to ovarian failure [1].
Iatrogenic Causes: Cancer treatments represent a significant iatrogenic cause, with the prevalence of POI among childhood cancer survivors ranging from 7.9% to 18.6% [1]. Alkylating agents (cyclophosphamide) and platinum-based drugs (cisplatin) are particularly gonadotoxic, causing follicle depletion through DNA damage, oxidative stress, and mitochondrial dysfunction [1]. Radiotherapy poses substantial risk, with even low doses (2 Gy) capable of destroying half of the ovarian follicle pool [1].
Environmental and Metabolic Factors: Environmental pollutants including phthalates, bisphenol A, pesticides, and tobacco have been associated with increased follicular atresia and accelerated ovarian aging [1]. Smoking has been consistently linked to POI risk, with studies showing a dose-dependent association and up to 2.75-fold elevated risk among smokers [1]. Classic galactosemia, a rare metabolic disorder, also predisposes to POI through toxic metabolite accumulation [1].
Mendelian Randomization: A Powerful Approach for Deconstructing Idiopathic POI
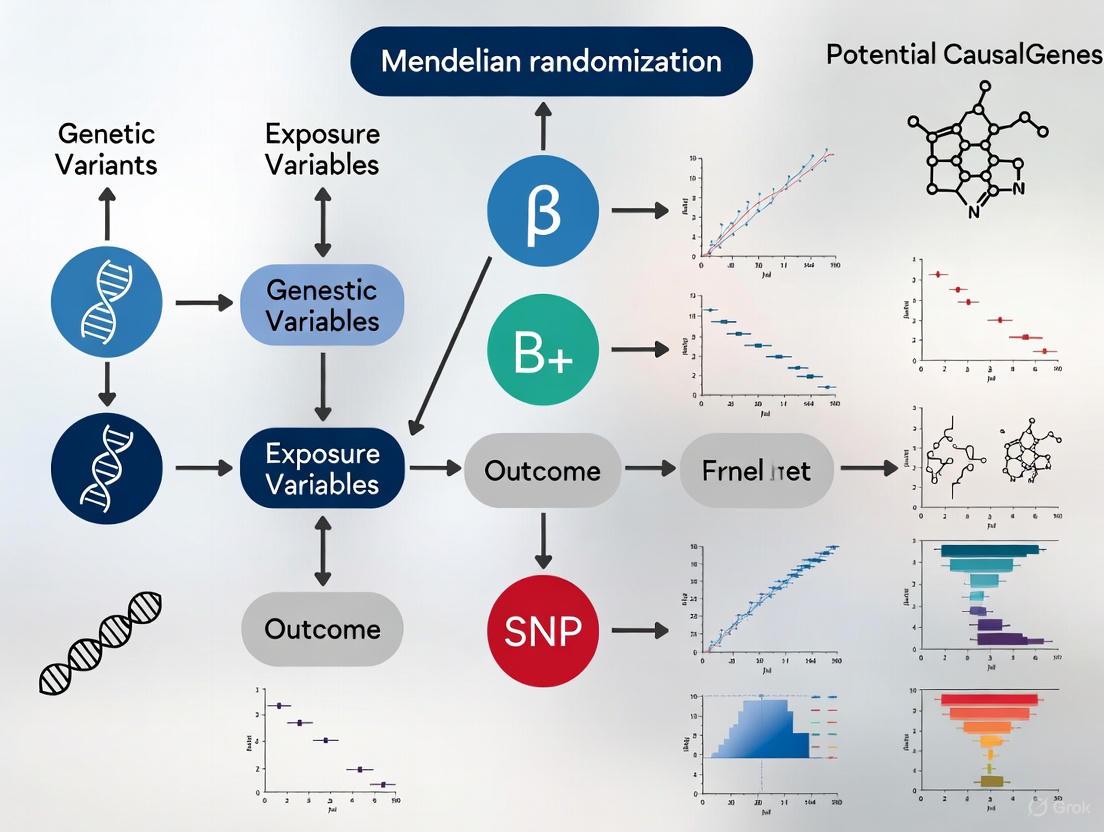
Mendelian randomization (MR) has emerged as a robust methodological framework for identifying causal genes and molecular pathways in POI, particularly for cases currently classified as idiopathic. MR uses genetic variants as instrumental variables to infer causal relationships between modifiable exposures or molecular traits (e.g., gene expression) and disease outcomes [5] [6] [7]. This approach minimizes confounding and avoids reverse causation, two major limitations of observational studies.
The core assumptions of MR are: (1) the genetic variants are strongly associated with the exposure; (2) the variants are independent of confounders; and (3) the variants affect the outcome only through the exposure [5] [7]. When applied to POI, MR integrates genome-wide association study (GWAS) data with expression quantitative trait loci (eQTL) data to test whether genetically predicted expression of specific genes has a causal effect on POI risk [6] [7].
Figure 1: Mendelian Randomization Workflow for POI Gene Discovery
Key MR Findings in POI
Recent MR studies have identified several genes with putative causal effects on POI:
FANCE (Fanconi Anemia Complementation Group E): MR and colocalization analyses strongly support FANCE as a causal gene for POI [6]. FANCE is involved in DNA repair through the Fanconi anemia pathway, and defects during primordial germ cell proliferation can lead to impaired cell division, reduced ovarian reserve, and POI [4].
RAB2A (Member RAS Oncogene Family): MR analyses identified RAB2A as significantly associated with reduced POI risk [6]. This gene is involved in autophagy regulation, a process crucial for oocyte survival and follicular development.
Additional Candidate Genes: A comprehensive MR analysis integrating multiple omics data identified non-invasive markers for POI warning, including three metabolites (sphinganine-1-phosphate, X-23636, 4-methyl-2-oxopentanoate), two circulating plasma proteins (fibroblast growth factor 23, neurotrophin-3), and 23 miRNAs [5].
Figure 2: Causal Pathways from Gene Expression to POI Risk Identified by MR
Experimental Protocols for MR Analysis in POI Research
Protocol 1: Two-Sample Mendelian Randomization
Purpose: To estimate the causal effect of gene expression on POI risk using summary-level GWAS and eQTL data from independent samples [5] [6] [7].
Procedure:
- Data Acquisition:
Instrumental Variable Selection:
- Identify single nucleotide polymorphisms (SNPs) associated with gene expression at genome-wide significance (P < 1×10⁻⁵) [5].
- Clump SNPs to ensure independence (linkage disequilibrium R² < 0.001 within 10,000 kb window) [5].
- Calculate F-statistic to assess instrument strength; retain instruments with F > 10 to avoid weak instrument bias [5].
MR Analysis:
- Perform inverse-variance weighted (IVW) MR as primary analysis method [5] [7].
- Conduct sensitivity analyses using MR-Egger, weighted median, and weighted mode methods [5].
- Apply false discovery rate (FDR) correction (FDR-adjusted P < 0.05) with odds ratio (OR) > 1.5 or < 0.5 considered statistically significant [5].
Sensitivity Analyses:
Purpose: To test whether the genetic association with POI is mediated by gene expression while distinguishing causality from linkage [6] [7].
Procedure:
- SMR Analysis:
Colocalization Analysis:
- Perform Bayesian colocalization using the coloc R package [6].
- Calculate posterior probabilities for five hypotheses:
- PP.H0: No association with either trait
- PP.H1: Association with gene expression only
- PP.H2: Association with POI only
- PP.H3: Association with both traits, different causal variants
- PP.H4: Association with both traits, same causal variant
- Define strong evidence of colocalization as PP.H3 + PP.H4 ≥ 0.8 [6].
Druggability Assessment:
- Query Online Mendelian Inheritance in Man (OMIM), DrugBank, Drug-Gene Interaction database (DGIdb), and Therapeutic Target Database (TTD) to evaluate potential for therapeutic targeting [6].
Table 3: Key Research Reagent Solutions for POI Mendelian Randomization Studies
| Resource Category | Specific Examples | Function and Application |
|---|---|---|
| GWAS Data Sources | FinnGen R11 (542 cases, 241,998 controls) [5] [6] | Provides summary statistics for POI genetic associations |
| eQTL Databases | GTEx V8 (Ovary: n=167; Whole Blood: n=670) [6], eQTLGen Consortium (n=31,684) [5] [6] | Source of genetic variants associated with gene expression |
| Analysis Tools | SMR software (v1.3.1) [6], coloc R package [6], TwoSample MR R package [5] | Perform MR, colocalization, and sensitivity analyses |
| Genetic Instruments | Cis-eQTL SNPs (P < 1×10⁻⁵, F > 10, R² < 0.001) [5] | Instrumental variables for causal inference |
| Annotation Databases | OMIM, ClinVar, gnomAD, CADD [2] | Assess variant pathogenicity and functional impact |
| Pathway Analysis | KEGG, GO, String database [5] | Biological interpretation of identified genes |
The integration of Mendelian randomization approaches with multi-omics data represents a powerful strategy for deconstructing the molecular basis of idiopathic POI. Current MR studies have already identified promising causal genes including FANCE and RAB2A, which implicate DNA repair mechanisms and autophagy regulation in POI pathogenesis. These findings not only advance our understanding of POI biology but also provide potential targets for future therapeutic interventions. As GWAS sample sizes expand and functional genomics resources become more comprehensive, MR approaches will continue to illuminate the genetic architecture of this complex condition, ultimately reducing the proportion of cases classified as idiopathic and enabling more personalized management strategies for affected women.
This application note details the integration of core biological processes—specifically folliculogenesis—with advanced Mendelian randomization (MR) methodologies to identify causal genetic factors and biomarkers for Premature Ovarian Insufficiency (POI). POI, the loss of ovarian function before age 40, affects approximately 3.7% of women globally, and its etiology remains complex and often unexplained [5]. A deep understanding of folliculogenesis provides the biological context for interpreting genetic discoveries, while MR offers a robust statistical framework to infer causality from genetic data, thereby informing drug target prioritization and the development of non-invasive diagnostic markers [5] [8] [9]. This protocol is designed for researchers and drug development professionals aiming to bridge the gap between ovarian biology and genetic epidemiology.
Biological Foundation: Folliculogenesis
Folliculogenesis is the protracted developmental process through which a primordial follicle matures into a Graafian follicle capable of ovulation. This process is fundamental to female fertility and forms the physiological basis for understanding POI.
Chronology and Key Stages
The journey from a primordial to a preovulatory follicle in humans requires nearly one year [10] [11]. This timeline can be divided into two main phases:
- Preantral (Gonadotropin-Independent) Phase: Lasting about 290 days, this phase encompasses the primordial, primary, and secondary follicle stages. Growth during this period is primarily regulated by autocrine/paracrine signals from the oocyte and surrounding somatic cells [10] [11].
- Antral (Gonadotropin-Dependent) Phase: Lasting approximately 60 days, this phase begins with antrum formation. The final 15-20 days of this phase involve the rapid growth of the dominant follicle selected during the late luteal phase of the menstrual cycle, a process critically dependent on FSH and LH [10] [11] [12].
Table 1: Key Stages of Human Folliculogenesis
| Stage | Diameter | Key Cellular Events | Primary Regulatory Mechanisms |
|---|---|---|---|
| Primordial | ~29 μm [11] | Oocyte arrested in diplotene; single layer of flattened granulosa cells; basal lamina [10]. | PTEN/PI3K/FOXO3 pathway maintains quiescence [10] [13]. Locally secreted factors (e.g., AMH, SDF-1) inhibit activation [10]. |
| Primary | Granulosa cells become cuboidal and proliferate; oocyte growth begins; zona pellucida formation [10]. | Oocyte-secreted factors (GDF9, BMP15) stimulate granulosa cell proliferation and FSHR expression [10] [13]. Kit ligand/KIT receptor interaction [10]. | |
| Secondary | Multiple layers of granulosa cells; formation of theca cell layer from surrounding stroma [11]. | Continued action of GDF9 and BMP15; onset of theca cell function [10]. | |
| Antral | 0.4 mm to >20 mm [11] | Formation of fluid-filled antrum; differentiation of granulosa into cumulus oophorus and mural layers; massive follicular growth. | FSH is essential for antrum formation and estrogen synthesis. LH stimulates androgen production in theca cells; LHR expression is detectable even in small antral follicles [10] [12]. |
Signaling Pathways in Early Follicle Activation
The transition of a primordial follicle from a quiescent to a growing primary follicle, known as recruitment or activation, is a critical checkpoint. Dysregulation of this process is a hypothesized mechanism for POI, as accelerated activation can prematurely deplete the ovarian reserve [10] [13]. The following diagram illustrates the key molecular pathways controlling this transition, integrating signals from the oocyte, granulosa cells, and the ovarian stroma.
Diagram 1: Molecular signaling in the primordial to primary follicle transition. Key pathways maintaining quiescence (red inhibitory arrows) and promoting activation (green arrows) are shown. Created with DOT language.
Mendelian Randomization in POI Research
Conceptual Framework and Workflow
Mendelian randomization is an instrumental variable method that uses genetic variants as proxies for modifiable exposures to assess causal relationships with outcomes [8] [9]. Its core strength lies in overcoming confounding and reverse causation, major limitations of observational studies, because genetic variants are randomly assorted at conception [8].
When applied to POI research, MR leverages large-scale Genome-Wide Association Study (GWAS) summary statistics to investigate the causal effect of various biomarkers, physiological traits, or lifestyle factors on POI risk. The following diagram outlines a typical multi-omics MR workflow for POI biomarker discovery.
Diagram 2: Integrated multi-omics Mendelian randomization workflow for POI research. Created with DOT language.
Core MR Assumptions and Their Application to POI
For MR findings to be valid, three core assumptions must be satisfied [8] [9]:
- Relevance: The genetic instrument must be robustly associated with the exposure of interest (e.g., a specific plasma protein level).
- Independence: The genetic instrument must not be associated with any confounders of the exposure-outcome relationship.
- Exclusion Restriction: The genetic instrument must affect the outcome (POI) only through the exposure, not via alternative pathways (no horizontal pleiotropy).
Integrated Application Note: A Multi-omics MR Protocol for POI
This protocol provides a detailed methodology for implementing the MR workflow to identify non-invasive biomarkers and causal genes for POI, as demonstrated in recent research [5].
Experimental Protocol: Two-Sample Mendelian Randomization
Objective: To assess the causal effect of a wide range of molecular traits on POI risk using summary-level GWAS data.
Data Sources:
- Outcome (POI): GWAS summary statistics from public databases (e.g., FinnGen R11 release: 542 cases, 241,998 controls) [5].
- Exposures: Summary statistics for various omics traits:
Step-by-Step Procedure:
- Instrumental Variable (IV) Selection:
- For each exposure dataset, extract single-nucleotide polymorphisms (SNPs) significantly associated with the trait at a genome-wide threshold (e.g., ( P < 1 \times 10^{-5} )) [5].
- Clump SNPs to ensure independence (linkage disequilibrium ( R^2 < 0.001 ) within a 10,000 kb window).
- Calculate the F-statistic for each SNP to ensure instrument strength (F > 10 is recommended to avoid weak instrument bias) [5].
Harmonization of Effects:
- Align the effect alleles of the exposure and outcome datasets for each selected SNP.
- Palindromic SNPs with intermediate allele frequencies should be excluded to avoid ambiguity.
MR Estimation:
- Perform Two-Sample MR analysis using the
TwoSampleMRpackage in R or similar. - Use the Inverse-Variance Weighted (IVW) method as the primary analysis for causal estimation [5].
- Apply supplementary methods ( MR-Egger, Weighted Median, Weighted Mode) to assess robustness.
- Perform Two-Sample MR analysis using the
Sensitivity Analysis:
- MR-Egger Intercept Test: Assess presence of directional pleiotropy (P < 0.05 suggests significant pleiotropy) [5].
- Cochran's Q Statistic: Test for heterogeneity among the causal estimates of individual SNPs (P < 0.05 indicates significant heterogeneity) [5].
- Leave-One-Out Analysis: Iteratively remove each SNP to determine if the causal effect is driven by a single influential variant.
Interpretation: A causal effect is supported if the IVW estimate yields an odds ratio (OR) significantly different from 1 (e.g., OR > 1.5 or < 0.5) with a false discovery rate (FDR)-adjusted ( P < 0.05 ) [5].
Protocol: Integrating Gene Expression via SMR
Objective: To test whether the effect of a genetic variant on POI is mediated by gene expression levels.
Procedure:
- Data Acquisition: Obtain expression Quantitative Trait Loci (eQTL) data from a relevant consortium (e.g., eQTLGen Consortium, 31,684 individuals) [5].
- SMR Analysis: Perform Summary-data-based MR (SMR) to test for association between gene expression and POI using top eQTLs as instruments.
- HEIDI Test: Conduct the heterogeneity in dependent instruments (HEIDI) test to distinguish linkage from pleiotropy. A result of ( P_{HEIDI} > 0.05 ) suggests the association is due to a shared causal variant (pleiotropy), supporting a causal link.
Key Findings from Recent Multi-omics MR for POI
A recent MR study identified several non-invasive markers for POI, summarized in the table below [5]. These findings exemplify the output of the described protocols.
Table 2: Exemplary Non-invasive Markers for POI Identified via MR
| Marker Category | Specific Identified Markers | Potential Functional Role |
|---|---|---|
| Metabolites | Sphinganine-1-phosphate, X-23636, 4-methyl-2-oxopentanoate | Involved in sphingolipid signaling and branched-chain amino acid metabolism [5]. |
| Plasma Proteins | Fibroblast growth factor 23 (FGF-23), Neurotrophin-3 (NT-3) | Regulation of phosphate metabolism, neuronal and ovarian development [5]. |
| MicroRNAs | miR-145-5p, miR-23a-3p, miR-221-3p, miR-146a-3p, and 19 others | Post-transcriptional regulators of genes in critical pathways like PI3K-Akt signaling and glutathione metabolism [5]. |
| Gut Microbiota | Faecalibacterium abundance | Butyrate-producing bacterium; may influence systemic inflammation and immune regulation [5]. |
| Hub Genes | ESR1, ERBB2, GART | Identified from protein-protein interaction networks; central to follicular development and folate metabolism [5]. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Resources for POI and Folliculogenesis Research
| Item/Category | Function/Application | Examples & Notes |
|---|---|---|
| GWAS Summary Data | Primary data for exposure and outcome in MR studies. | FinnGen (POI), UK Biobank, eQTLGen Consortium (eQTLs), GWAS Catalog [5] [14]. Publicly accessible. |
| MR Software & Packages | Statistical analysis of causal inference. | TwoSampleMR (R), MR-Base platform, SMR software [5] [9]. |
| Pathway Analysis Tools | Functional annotation of identified genes/miRNAs. | KEGG, String database (PPI networks), Cytoscape, miEAA (for miRNA pathway enrichment) [5]. |
| Cell & Animal Models | Functional validation of candidate genes/pathways. | Mouse models (e.g., for FIGLA, FOXL2, PTEN mutations) [10]. Bovine oocyte model for human extrapolation [13]. |
| Key Antibodies | Detection of protein expression in ovarian tissues. | Anti-LHR monoclonal antibody (e.g., 3B5) for detecting LHR in theca cells of preantral follicles [12]. |
| Recombinant Proteins & Inhibitors | Manipulating signaling pathways in vitro. | Recombinant GDF9, BMP15; PTEN inhibitors (e.g., bpV(HOpic)); PI3K/AKT pathway modulators [10] [13]. |
The integration of detailed folliculogenesis biology with the causal inference power of Mendelian randomization creates a powerful paradigm for POI research. The protocols outlined here provide a roadmap for identifying and validating causal biomarkers and genes, offering direct paths to clinical translation through non-invasive diagnostics and prioritized drug targets. This multi-omics, genetics-driven approach significantly advances our ability to understand, predict, and potentially intervene in the complex etiology of Premature Ovarian Insufficiency.
Primary Ovarian Insufficiency (POI) is a major cause of female infertility, characterized by the cessation of ovarian function before age 40, affecting approximately 1-3.7% of women [4]. This application note details the methodologies and findings from a landmark large-scale whole-exome sequencing (WES) study that systematically identified pathogenic variants in 59 known POI-causative genes. The study by et al. published in Nature Medicine (2023) represents the largest WES study in patients with POI to date, providing unprecedented insights into the genetic architecture of this heterogeneous condition [2]. Within the broader context of Mendelian randomization research for POI, which uses genetic variants as instrumental variables to infer causal relationships, the robust identification of pathogenic variants in known genes is a critical first step. This establishes a foundation for subsequent causal inference and drug target validation by pinpointing genuine genetic risk factors free from confounding and reverse causation biases inherent in observational studies [15] [16].
The study cohort comprised 1,030 unrelated women with POI, including 120 with primary amenorrhea (PA) and 910 with secondary amenorrhea (SA). All participants underwent WES, and variant pathogenicity was evaluated according to American College of Medical Genetics and Genomics (ACMG) guidelines [2].
Table 1: Overall Genetic Diagnostic Yield in the POI Cohort
| Category | Number of Patients | Percentage of Cohort |
|---|---|---|
| Total POI patients | 1,030 | 100% |
| Patients with P/LP variants in known genes | 193 | 18.7% |
| Patients with monoallelic variants | 155 | 15.0% |
| Patients with biallelic variants | 24 | 2.3% |
| Patients with multiple heterozygous variants | 14 | 1.4% |
Table 2: Distribution of 195 P/LP Variants by Type and Functional Consequence
| Variant Type | Number of Variants | Percentage |
|---|---|---|
| Loss-of-Function (LoF) | 108 | 55.4% |
| Frameshift indels | 38 | 19.5% |
| Nonsense | 44 | 22.6% |
| Canonical splice site | 23 | 11.8% |
| Start-loss | 3 | 1.5% |
| Missense | 81 | 41.5% |
| In-frame indels | 4 | 2.1% |
| Splice region | 2 | 1.0% |
Table 3: Top Contributing Genes and Associated Biological Pathways
| Gene Symbol | Patients with P/LP Variants (n) | Primary Amenorrhea (n=120) | Secondary Amenorrhea (n=910) | Key Biological Pathway |
|---|---|---|---|---|
| NR5A1 | 11 | 1 (0.8%) | 10 (1.1%) | Steroidogenesis / Folliculogenesis |
| MCM9 | 11 | 2 (1.7%) | 9 (1.0%) | Meiosis / DNA Repair |
| EIF2B2 | 10 | 1 (0.8%) | 9 (1.0%) | Translation / Metabolism |
| HFM1 | 9 | 2 (1.7%) | 7 (0.8%) | Meiosis / Homologous Recombination |
| BRCA2 | 8 | 0 (0%) | 8 (0.9%) | DNA Damage Repair |
| FSHR | 7 | 5 (4.2%) | 2 (0.2%) | Folliculogenesis / Signaling |
The study identified 195 pathogenic or likely pathogenic (P/LP) variants across 59 known POI-causative genes, contributing to 193 (18.7%) of the 1,030 cases [2]. Most cases (155/193, 80.3%) involved monoallelic variants, while biallelic and multiple heterozygous variants accounted for 12.4% and 7.3%, respectively. Genes involved in meiosis and DNA repair constituted the largest functional group, underlying nearly half (48.7%) of the genetically explained cases [2].
Experimental Protocols for Variant Identification and Validation
Patient Cohort Selection and Diagnostic Criteria
- Inclusion Criteria: The study recruited 1,030 unrelated women diagnosed with POI according to the European Society of Human Reproduction and Embryology (ESHRE) guidelines.
- Oligomenorrhea or amenorrhea for at least 4 months before 40 years of age.
- Elevated follicle-stimulating hormone (FSH) level >25 IU/L on two occasions more than 4 weeks apart [2].
- Exclusion Criteria: Patients with chromosomal abnormalities, FMR1 premutations, or known non-genetic causes (autoimmune diseases, ovarian surgery, chemotherapy, radiotherapy) were excluded [2].
- Control Cohort: For association analysis, an in-house control cohort of 5,000 unrelated individuals from the HuaBiao project was used, generated with the same exome capture kit to minimize technical bias [2].
Whole Exome Sequencing (WES) and Bioinformatic Analysis
The following protocol details the key steps for generating and analyzing WES data, as described across multiple studies [17] [2] [18].
Step 1: DNA Extraction and Library Preparation
- Extract genomic DNA from patient blood or other appropriate tissues using standard methods (e.g., Qiagen kits) [17].
- Prepare exome sequencing libraries using commercial exome capture kits (e.g., Agilent SureSelect, Roche NimbleGen VCRome) [17].
Step 2: Whole Exome Sequencing
- Perform high-throughput sequencing on an Illumina platform (e.g., HiSeq 2500, HiSeq 2000) to generate paired-end reads [17].
Step 3: Sequence Alignment and Variant Calling
- Align sequencing reads (FASTQ files) to the human reference genome (GRCh37/hg19) using the Burrows-Wheeler Aligner (BWA-MEM) [17].
- Process aligned BAM files by marking duplicates and performing base quality recalibration using software like Sentieon or the Genome Analysis Toolkit (GATK) [17].
- Call germline variants (SNVs and indels) using a joint-calling approach across all samples with a variant caller such as Sentieon Haplotyper or GATK HaplotypeCaller to produce a multi-sample VCF file [17] [2].
Step 4: Variant Quality Control and Filtration
- Apply stringent quality control filters to remove low-quality variants and technical artifacts.
- Annotate variants using tools like ANNOVAR and Ensembl VEP to predict functional consequences [19] [2].
- Filter out common variants by retaining only those with a minor allele frequency (MAF) < 0.01 in population databases (e.g., gnomAD) and the in-house control cohort [2].
Step 5: Pathogenicity Assessment and Prioritization
- Classify variants according to ACMG/AMP guidelines to identify Pathogenic (P) and Likely Pathogenic (LP) variants [2].
- Utilize in silico prediction tools (e.g., CADD, SIFT, PolyPhen-2) to support deleteriousness predictions. In the landmark study, 94.4% of P/LP variants had a CADD score >20 [2].
- Prioritize variants in known POI-causative genes (95 genes were initially screened) [2].
Step 6: Validation of Candidate Variants
- Confirm prioritized P/LP variants, especially those classified as Variants of Uncertain Significance (VUS) that are upgraded based on functional evidence, using an independent method such as Sanger sequencing [17] [2].
- For biallelic variants, confirm the in trans configuration (on opposite alleles) through T-clone sequencing or long-read technologies (e.g., 10x Genomics) [2].
Functional Validation of Variants of Uncertain Significance (VUS)
The study highlighted the importance of functional assays to resolve VUS.
- Approach: Select VUS in key POI genes for experimental validation.
- Protocol: The researchers functionally validated 75 VUS from seven genes involved in homologous recombination repair (BLM, HFM1, MCM8, MCM9, MSH4, RECQL4) and folliculogenesis (NR5A1).
- Outcome: Of these, 55 variants were confirmed to be deleterious, and 38 were subsequently upgraded from VUS to Likely Pathogenic (LP), significantly increasing the diagnostic yield [2]. Specific assay methodologies (e.g., for assessing DNA repair function or transcriptional activity) should be tailored to the gene's known function.
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents and Tools for POI Genetic Studies
| Item | Function/Application | Example Kits/Software (from studies) |
|---|---|---|
| Exome Capture Kits | Target enrichment for sequencing | Agilent SureSelect, Roche NimbleGen VCRome 2.1 [17] |
| NGS Platform | High-throughput DNA sequencing | Illumina HiSeq 2500/2000 [17] |
| Alignment Tool | Map sequencing reads to reference genome | BWA-MEM [17] |
| Variant Caller | Identify genetic variants from aligned data | Sentieon Haplotyper, GATK HaplotypeCaller [17] |
| Variant Annotator | Predict functional impact of variants | ANNOVAR, Ensembl VEP [19] [2] |
| Population Database | Filter common polymorphisms | gnomAD [19] [2] |
| Pathogenicity Predictor | In silico assessment of variant deleteriousness | CADD, SIFT, PolyPhen-2 [2] |
| Sanger Sequencing | Independent validation of candidate variants | Standard dye-terminator methods [17] [18] |
Integration with Mendelian Randomization Research
The precise identification of pathogenic variants in POI genes, as detailed in this protocol, provides the fundamental genetic associations required for robust Mendelian randomization (MR) analyses. In MR, genetic variants serve as instrumental variables to proxy the lifelong effect of perturbing a drug target, thereby inferring causal effects on health and disease outcomes [15] [16].
- From Association to Causation: P/LP variants in genes like MCM9 or NR5A1 are not merely associated with POI; their deleterious nature suggests a direct causal role in the disease pathway. This makes them high-quality instruments for MR [16].
- Informing Drug Development: MR can use these genetic instruments to predict the efficacy and potential on-target side effects of modulating a gene's protein product. For example, genetic variants that impair the function of a protein and cause POI would predict that a therapeutic inhibitor of that protein could negatively impact ovarian reserve [15] [16]. Conversely, identifying protective LoF variants can highlight promising new drug targets.
- Addressing Pitfalls: The rigorous variant filtering and functional validation protocols described here help mitigate key MR assumptions violations, such as pleiotropy (where a genetic variant influences multiple traits), by ensuring the variant's effect is specific to the gene and pathway of interest [16].
This application note outlines a comprehensive and robust framework for identifying pathogenic variants in POI-causative genes, as demonstrated by a landmark study that achieved an 18.7% molecular diagnostic rate. The integration of large cohort WES, stringent bioinformatic filtering, ACMG classification, and functional validation provides a high-yield genetic testing protocol. These findings and methods are instrumental for clinical diagnostics, genetic counseling, and for building a genetically validated foundation for Mendelian randomization studies aimed at de-risking and accelerating drug development for ovarian infertility.
Genetic Distinctions: Comparing Primary vs. Secondary Amenorrhea Profiles presents a systematic framework for investigating the genetic architectures of primary and secondary amenorrhea within research on Mendelian randomization (MR) for primary ovarian insufficiency (POI) causal genes. Amenorrhea, the absence of menstruation, is categorized as primary amenorrhea (PA) when menarche has not occurred by age 15 or within three years of thelarche, and secondary amenorrhea (SA) when established menses cease for ≥3 months in women with previous regular cycles or ≥6 months in those with prior irregular cycles [20] [21] [22]. Understanding the genetic underpinnings distinguishing these presentations is critical for elucidating POI pathogenesis and developing targeted therapeutic interventions for researchers and drug development professionals.
The application of Mendelian randomization principles offers a powerful approach to infer causality in epidemiological studies by utilizing genetic variants as instrumental variables to examine the effect of modifiable risk factors on disease outcomes [23]. Within reproductive genetics, MR studies have begun to identify causal relationships between genetic predispositions, altered reproductive traits, and subsequent disease risks, providing a robust methodological foundation for dissecting the genetic causality in POI and related amenorrhea phenotypes [23].
Clinical Definitions and Etiological Frameworks
Diagnostic Criteria and Classification
The clinical distinction between primary and secondary amenorrhea forms the foundation for etiological investigation and genetic analysis. The diagnostic frameworks and epidemiological characteristics are summarized in Table 1.
Table 1: Clinical Definitions and Epidemiological Patterns of Primary and Secondary Amenorrhea
| Parameter | Primary Amenorrhea | Secondary Amenorrhea |
|---|---|---|
| Definition | Absence of menarche by age 15 years or within 3 years of thelarche [20] [21] | Cessation of menses for ≥3 months (previously regular cycles) or ≥6 months (previously irregular cycles) [20] [21] |
| Prevalence | Rare (<1%) [22] | Approximately 3-4% (excluding pregnancy, lactation, menopause) [22] |
| Common Etiologies | Gonadal dysgenesis (e.g., Turner syndrome), Müllerian anomalies, constitutional delay [20] [22] | Functional hypothalamic amenorrhea, PCOS, hyperprolactinemia, POI [20] [21] |
| Typical Age at Presentation | Adolescence (13-18 years) [20] | Reproductive years (variable) [20] |
| Frequently Implicated Genetic Loci | Chromosomal abnormalities (e.g., 45,X), SRY genes, Müllerian development genes [20] | POI-associated genes (e.g., FMR1 premutation), GnRH neuronal function genes [20] [24] |
Etiological Pathways and Genetic Correlates
The pathophysiological mechanisms underlying amenorrhea can be categorized according to disruptions within specific components of the hypothalamic-pituitary-ovarian (HPO) axis and genital outflow tract, each with distinct genetic associations:
Outflow Tract Abnormalities: Predominant in PA, including Müllerian agenesis (Mayer-Rokitansky-Küster-Hauser syndrome) and complete androgen insensitivity syndrome (CAIS) [20]. These conditions frequently involve genetic mutations affecting embryonic development of reproductive structures [20] [22].
Ovarian Dysfunction: Encompasses both PA and SA, with primary ovarian insufficiency (POI) representing a critical intersection point. POI is defined as hypergonadotropic hypogonadism before age 40 [20] [23]. Genetic etiologies include chromosomal abnormalities (e.g., Turner syndrome), FMR1 premutations, and various single gene disorders [20].
Hypothalamic/Pituitary Disorders: More common in SA, including functional hypothalamic amenorrhea (FHA) and hyperprolactinemia [20] [21]. Recent evidence suggests genetic susceptibility in FHA through rare sequence variants in genes associated with gonadotropin-releasing hormone (GnRH) neuronal function [24].
Other Endocrine Disorders: Particularly polycystic ovary syndrome (PCOS), a common cause of SA with strong heritability components [20].
Table 2: Genetic Associations in Amenorrhea Etiologies
| Etiological Category | Example Conditions | Key Genetic Associations |
|---|---|---|
| Gonadal Disorders | Turner syndrome (45,X) | Chromosomal aneuploidy [20] |
| Primary ovarian insufficiency | FMR1 premutation, chromosomal abnormalities, autoimmune polyglandular syndromes [20] | |
| Pure gonadal dysgenesis (Sweyer syndrome) | 46,XY SRY gene mutations [20] | |
| Outflow Tract Abnormalities | Müllerian agenesis | Unknown; often sporadic [20] |
| Complete androgen insensitivity syndrome (CAIS) | Androgen receptor gene mutations [20] | |
| Hypothalamic/Pituitary Disorders | Functional hypothalamic amenorrhea | Rare sequence variants in GnRH-associated genes [24] |
| Kallmann syndrome | KAL1, FGFR1, PROKR2, PROK2 genes [20] |
Mendelian Randomization Approaches in Amenorrhea Research
Methodological Framework
Mendelian randomization represents a sophisticated epidemiological approach that utilizes genetic variants as instrumental variables to infer causal relationships between modifiable risk factors and health outcomes [23]. This method relies on three core assumptions: (1) the genetic variant is robustly associated with the exposure, (2) the variant is independent of confounders, and (3) the variant influences the outcome only through the exposure [23].
In the context of amenorrhea research, MR designs can be implemented through several approaches:
- One-sample MR: Utilizes a single cohort for both genetic instrument derivation and outcome assessment [23].
- Two-sample MR: Employs two independent cohorts, typically yielding more conservative estimates with lower false-positive rates [23].
- Multivariable MR (MVMR): Simultaneously analyzes direct causal effects of multiple correlated exposures [23].
- Mediation MR: Identifies factors that mediate the relationship between exposure and outcome [23].
Application to Amenorrhea and POI Research
MR studies have elucidated causal relationships between reproductive traits and subsequent disease risks, providing a template for investigating genetic causality in amenorrhea. Key findings with methodological relevance include:
- Early age at menarche demonstrates causal association with early age at natural menopause [23].
- Higher childhood BMI exhibits causal relationships with early menarche and early menopause [23].
- Educational attainment shows causal association with age at natural menopause, with longer education genetically predisposing to later menopause [23].
These established relationships demonstrate the utility of MR for investigating causal pathways in reproductive disorders, including the genetic distinctions between primary and secondary amenorrhea presentations.
Experimental Protocols for Genetic Analysis
Protocol 1: Mendelian Randomization Analysis for Causal Inference
Objective: To implement a two-sample MR analysis examining causal effects of genetic predispositions to reproductive traits on amenorrhea risk.
Materials:
- Genome-wide association study (GWAS) summary statistics for exposure traits (e.g., age at menarche, BMI, educational attainment)
- GWAS summary statistics for amenorrhea outcomes (primary vs. secondary)
- MR analysis software (e.g., TwoSampleMR R package, MR-Base platform)
Procedure:
- Instrument Selection: Identify single nucleotide polymorphisms (SNPs) strongly associated (p < 5 × 10⁻⁸) with exposure traits from large-scale GWAS meta-analyses.
- Harmonization: Align effect alleles for exposure and outcome datasets, ensuring consistent effect direction.
- MR Analysis Implementation:
- Perform inverse-variance weighted (IVW) method as primary analysis
- Conduct sensitivity analyses (MR-Egger, weighted median, MR-PRESSO)
- Assess heterogeneity using Cochran's Q statistic
- Test for horizontal pleiotropy via MR-Egger intercept
- Validation: Apply multivariable MR to account for correlated exposures and mediation analysis to identify potential intermediary pathways.
Protocol 2: Rare Variant Burden Testing in Amenorrhea Cohorts
Objective: To identify enrichment of rare sequence variants (RSVs) in genes associated with isolated hypogonadotropic hypogonadism (IHH) in women with functional hypothalamic amenorrhea.
Materials:
- Case cohort: Women with documented FHA (n = 106) [24]
- Control cohort: Healthy postmenopausal women with normal reproductive history (n = 468) [24]
- Exome sequencing data for all participants
- Variant annotation pipeline (e.g., SnpEff, VEP)
- Rare variant burden testing software (e.g., SKAT-O, Burden tests)
Procedure:
- Case Ascertainment: Recruit women with FHA according to standardized criteria: ≥6 months amenorrhea with documented risk factors (energy deficit, exercise, stress) and exclusion of other endocrine disorders [24].
- Control Selection: Identify postmenopausal women aged 45-65 years with spontaneous menarche history and no amenorrhea excluding pregnancy [24].
- Gene Panel Definition: Curate list of IHH-associated genes (e.g., GNRHR, KISS1R, TAC3, TACR3) based on established literature [24].
- Variant Filtering:
- Quality control: Remove variants with call rate <95% or Hardy-Weinberg equilibrium p < 1 × 10⁻⁶
- Frequency filtering: Retain variants with minor allele frequency <0.1% in reference populations
- Functional prediction: Prioritize loss-of-function and deleterious missense variants
- Burden Analysis: Perform gene-based rare variant association tests comparing FHA cases versus controls, adjusting for appropriate covariates.
Protocol 3: POI Gene Panel Sequencing and Validation
Objective: To identify and validate pathogenic variants in known POI-associated genes across primary and secondary amenorrhea presentations.
Materials:
- DNA samples from amenorrhea cohorts (PA with hypergonadotropic hypogonadism; SA with POI)
- Targeted sequencing panel for POI genes (e.g., FMR1, BMP15, FOXL2, NR5A1)
- Sanger sequencing reagents for validation
- Functional assay systems (e.g., in vitro granulosa cell models)
Procedure:
- Patient Stratification: Categorize amenorrhea patients by:
- Primary vs. secondary presentation
- Hormonal profile (hypogonadotropic vs. hypergonadotropic)
- Associated features (e.g., syndromic findings)
- Targeted Sequencing: Perform next-generation sequencing of POI gene panel with minimum 100x coverage.
- Variant Prioritization:
- Filter against population frequency databases (gnomAD, 1000 Genomes)
- Annotate functional impact using multiple prediction algorithms
- Classify according to ACMG/AMP guidelines
- Segregation Analysis: Confirm variant segregation with phenotype in available family members.
- Functional Validation:
- For missense variants: Express mutant proteins in cell culture systems
- For putative loss-of-function variants: Perform minigene splicing assays or CRISPR-edited cell lines
- Assess impact on granulosa cell function, apoptosis, and steroidogenesis
Visualization of Genetic Pathways and Analytical Frameworks
Genetic Susceptibility to Functional Hypothalamic Amenorrhea
Figure 1: Genetic-Environmental Interplay in Functional Hypothalamic Amenorrhea Pathogenesis. Rare sequence variants in genes associated with isolated hypogonadotropic hypogonadism increase susceptibility to developing amenorrhea in response to environmental stressors through dysregulation of the hypothalamic-pituitary-adrenal (HPA) axis and subsequent gonadotropin-releasing hormone (GnRH) neuronal dysfunction [24].
Mendelian Randomization Framework for Amenorrhea Research
Figure 2: Mendelian Randomization Framework for Causal Inference in Amenorrhea Research. The MR approach utilizes genetic variants as instrumental variables to infer causality between exposures (e.g., reproductive traits) and amenorrhea outcomes, under three core assumptions that minimize confounding [23].
Diagnostic Algorithm for Primary vs. Secondary Amenorrhea
Figure 3: Genetic Evaluation Algorithm for Primary and Secondary Amenorrhea. The diagnostic approach integrates clinical presentation with targeted genetic testing, directing specific genetic analyses based on initial clinical and biochemical findings [20] [21] [22].
Research Reagent Solutions
Table 3: Essential Research Reagents for Amenorrhea Genetic Studies
| Reagent/Category | Specific Examples | Research Application |
|---|---|---|
| Genetic Analysis Tools | Exome sequencing kits | Comprehensive variant detection across coding regions [24] |
| Targeted gene panels (POI, IHH genes) | Focused analysis of amenorrhea-associated genes [24] | |
| GWAS arrays (Illumina, Affymetrix) | Genome-wide association studies for variant discovery [23] | |
| Bioinformatic Resources | Variant annotation pipelines (SnpEff, VEP) | Functional prediction of genetic variants [24] |
| MR-Base platform, TwoSampleMR | Mendelian randomization analysis [23] | |
| gnomAD, 1000 Genomes | Population frequency databases for variant filtering [24] | |
| Functional Validation Assays | Granulosa cell culture systems | In vitro modeling of ovarian dysfunction [20] |
| GnRH neuronal cell models | Study of hypothalamic function [24] | |
| CRISPR-Cas9 gene editing | Functional characterization of candidate variants [24] | |
| Clinical Assessment Tools | ELISA/Luminex hormone assays | FSH, LH, estradiol, AMH quantification [20] [21] |
| Pelvic ultrasound | Assessment of ovarian morphology and uterine development [21] [22] | |
| Karyotyping/CNV analysis | Detection of chromosomal abnormalities [20] [22] |
The genetic distinctions between primary and secondary amenorrhea profiles provide critical insights for advancing Mendelian randomization applications in POI causal gene research. While primary amenorrhea often involves severe developmental genetic disorders and chromosomal abnormalities, secondary amenorrhea frequently presents more subtle genetic susceptibilities that interact with environmental factors. The experimental frameworks and analytical protocols presented herein enable systematic investigation of these genetic architectures, accelerating the identification of validated therapeutic targets for drug development pipelines. Through continued application of these approaches, researchers can elucidate the causal genetic pathways in amenorrhea, ultimately enabling personalized interventions based on individual genetic profiles.
The study of Premature Ovarian Insufficiency (POI) has traditionally focused on monogenic causes, where pathogenic variants in a single gene result in large physiological effects. However, most cases of POI, like many other complex diseases, result from the cumulative effects of multiple genetic variants and environmental factors [25]. In such polygenic diseases, each genetic variant usually confers only a small individual effect, making genetic studies comparatively more challenging than for monogenic disorders [25]. The emerging understanding that POI has a significant polygenic component represents a paradigm shift in how researchers approach its etiology and pathogenesis.
Polygenic Risk Scores (PRS) have emerged as a powerful quantitative tool to measure an individual's genetic susceptibility to complex diseases like POI. A PRS is calculated as the weighted sum of all risk alleles an individual carries for a specific trait, with weights proportional to each allele's effect size derived from genome-wide association studies (GWAS) [26]. This approach integrates the effects of numerous genetic variants across the genome, providing a comprehensive view of an individual's genetic risk profile that moves beyond single-gene determinants. For POI, which affects approximately 1% of the female population and leads to infertility and increased long-term health risks, understanding this polygenic architecture is crucial for advancing predictive, preventive, and therapeutic strategies [27] [28].
Polygenic Risk Scores: Quantifying Cumulative Genetic Burden
Principles and Calculation of Polygenic Risk Scores
The construction of Polygenic Risk Scores relies on summary data from large-scale genome-wide association studies (GWAS). In a GWAS, millions of genetic variants, typically single nucleotide polymorphisms (SNPs), are tested for association with a trait or disease across the genome [25] [29]. SNPs that show statistically significant associations are identified, along with their effect sizes (beta coefficients or odds ratios) and measures of statistical significance (p-values) [29]. The basic formula for calculating a PRS for an individual is:
PRS = Σ (βi × Gi)
Where βi is the effect size of the i-th SNP, and Gi is the genotype of the individual for that SNP (typically coded as 0, 1, or 2 copies of the effect allele) [26]. To ensure robust PRS calculation, several quality control steps are essential, including filtering for genome-wide significant variants (typically p < 5×10⁻⁸), accounting for linkage disequilibrium (LD) to select independent variants, and using an independent LD reference panel [30]. More advanced methods like PRS-CS (Continuous Shrinkage) apply Bayesian shrinkage to effect sizes, making them robust across diverse genetic architectures and improving predictive accuracy compared to traditional clumping and thresholding approaches [30].
PRS Performance and Clinical Utility in Complex Diseases
Evidence from large-scale studies demonstrates the significant influence of PRS on disease risk across multiple conditions. A comprehensive assessment of 32 complex diseases in the UK Biobank revealed that higher PRS led to greater incident risk, with hazard ratios (HR) ranging from 1.07 for panic/anxiety disorder to 4.17 for acute pancreatitis [30]. The effect was more pronounced in early-onset cases for many diseases, increasing by 52.8% on average. Specifically for heart failure, the early-onset risk associated with PRS (HR = 3.02) was roughly twice that of late-onset risk (HR = 1.48) [30].
Individuals in the top 2.5% of the PRS distribution exhibited varying degrees of elevated risk, corresponding to a more than five times greater risk on average compared to those with average PRS (20-80%) [30]. When incorporated into clinical risk prediction models, PRS provided additional value, causing an average improvement of 6.1% in prediction accuracy. The predictive accuracy was particularly higher for early-onset cases of 11 diseases, with heart failure showing the most significant improvement (37.5%) when PRS was added to the prediction model [30].
Table 1: Performance of Polygenic Risk Scores Across Selected Complex Diseases
| Disease | Hazard Ratio (HR) | Early-onset vs Late-onset HR Difference | C-index Improvement with PRS |
|---|---|---|---|
| Acute Pancreatitis | 4.17 (95% CI: 4.03-4.31) | Not reported | Not reported |
| Heart Failure | 2.15 (95% CI: 2.10-2.20) | +104% (Early: 3.02 vs Late: 1.48) | +37.5% |
| Panic/Anxiety Disorder | 1.07 (95% CI: 1.06-1.08) | Not reported | Not reported |
| Type 2 Diabetes | Not reported | Not reported | ~6.1% (average across diseases) |
Mendelian Randomization: Establishing Causal Relationships in POI
Principles and Assumptions of Mendelian Randomization
Mendelian Randomization (MR) is an epidemiological method that uses genetic variants as instrumental variables to investigate causal relationships between modifiable exposures and outcomes [31] [32]. The approach relies on three core assumptions: (1) the genetic variants must be strongly associated with the exposure (relevance assumption); (2) the genetic variants should not be associated with confounders of the exposure-outcome relationship (independence assumption); and (3) the genetic variants should affect the outcome only through the exposure, not through alternative pathways (exclusion restriction) [31]. Because alleles are inherited randomly at conception and cannot be modified by disease, MR estimates are resistant to bias from reverse causation and largely independent of environmental and lifestyle influences that often confound traditional observational studies [31].
The MR approach can be likened to a naturally randomized trial, where genetic variation serves as the randomization mechanism [32]. This is particularly valuable for investigating POI etiology, where randomized controlled trials are often not feasible or ethical. MR studies can be conducted using either one-sample or two-sample designs. In one-sample MR, both the instrument-exposure and instrument-outcome associations are estimated in the same cohort, while two-sample MR uses independent cohorts for these estimates, generally offering better generalizability [25] [31].
Application of MR in POI Research: Inflammatory Cytokines as Causal Factors
Recent MR studies have provided valuable insights into the causal relationships between inflammatory cytokines and POI. One investigation used genetic instruments for 91 inflammation-related proteins derived from 14,824 European participants and POI summary statistics from the FinnGen consortium (424 cases and 118,796 controls) [33]. The study employed multiple MR methods, with the inverse-variance weighted (IVW) method serving as the primary approach, supplemented by MR-Egger, weighted median, and other sensitivity analyses.
The findings revealed that specific inflammatory proteins exert protective effects against POI, while others increase risk. CXCL10 and CX3CL1 were identified as potentially protective, whereas IL-18R1, IL-18, MCP-1, and CCL28 were associated with increased POI risk [33]. Additional analyses highlighted protective effects of IL-17C, TRANCE, uPA, LAP TGF-β1, and CXCL9, along with risk proteins including TNFSF14, CD40, IL-24, ARTN, LIF-R, and IL-2RB [33]. Experimental validation in a POI cell model (KGN cells treated with cyclophosphamide) confirmed significant changes in MCP-1/CCL2, TGFB1, ARTN, and LIFR, which were found to converge in the oncostatin M signaling pathway [33].
A separate MR study focusing on inflammatory cytokines and POI identified CCL19, IL10, IL17A, and CCL7 as potentially protective against POI development, while IL-33 demonstrated a harmful association, possibly through its role in amplifying inflammatory processes that compromise ovarian function [27]. These findings collectively support the notion that immunomodulatory treatments might be viable approaches for preventing and managing POI.
Table 2: Causal Effects of Inflammatory Cytokines on POI Identified Through Mendelian Randomization
| Inflammatory Cytokine | Effect on POI Risk | MR Method | Potential Mechanism |
|---|---|---|---|
| CXCL10, CX3CL1 | Protective | IVW, Wald ratio | Anti-inflammatory signaling |
| IL-18, IL-18R1, MCP-1, CCL28 | Risk-increasing | IVW | Pro-inflammatory pathways |
| IL-17C, TRANCE, uPA, LAP TGF-β1 | Protective | Wald ratio | Immune regulation |
| IL-10, IL-17A, CCL7, CCL19 | Protective | IVW, MR-Egger | Anti-inflammatory effects |
| IL-33 | Risk-increasing | IVW | Amplification of inflammatory processes |
Experimental Protocols for Polygenic Risk and Mendelian Randomization Studies
Protocol for Polygenic Risk Score Calculation and Validation
Sample Preparation and Quality Control: Begin with genomic data from a representative cohort of cases and controls. Perform standard quality control procedures including filtering for call rate (>98%), Hardy-Weinberg equilibrium (p > 1×10⁻⁶), and minor allele frequency (>1%). Calculate principal components to account for population stratification [30].
PRS Calculation Using PRS-CS Method:
- Obtain GWAS summary statistics for the trait of interest from large consortium data.
- Use an external linkage disequilibrium reference panel such as the 1000 Genomes Project (n = 503 European samples) [30].
- Apply the PRS-CS auto option, which uses a Bayesian regression framework with a continuous shrinkage prior to effect sizes across the genome.
- Generate posterior effect sizes for all SNPs, which are more robust across diverse genetic architectures.
- Calculate individual PRS as the sum of allele counts weighted by these posterior effect sizes.
Validation and Assessment:
- Standardize PRS to mean = 0 and unit variance for association analyses.
- Evaluate association between PRS and disease status using regression models, adjusting for principal components and other relevant covariates.
- Assess predictive performance using measures such as the C-index (concordance index) and net reclassification improvement (NRI) [30].
- For comparative purposes, calculate PRS using alternative methods such as pruning and thresholding (P+T) to confirm superior performance of PRS-CS.
Protocol for Two-Sample Mendelian Randomization Analysis
Instrument Selection:
- Identify genetic instruments for the exposure (e.g., inflammatory cytokines) from published GWAS summary statistics.
- Select SNPs associated with the exposure at genome-wide significance (p < 5×10⁻⁸).
- Clump SNPs to ensure independence using linkage disequilibrium criteria (e.g., r² < 0.001 within 10,000 kb window) [33].
- Calculate F-statistics for each instrument to assess strength, excluding weak instruments (F < 10) to avoid bias [33].
MR Analysis Implementation:
- Obtain outcome (POI) summary statistics from an independent source (e.g., FinnGen consortium).
- Harmonize exposure and outcome datasets to ensure effect alleles correspond to the same strand.
- Perform primary analysis using the inverse-variance weighted (IVW) method under a random-effects model [33] [27].
- Conduct sensitivity analyses using multiple methods:
- MR-Egger regression to test for directional pleiotropy
- Weighted median estimator for consistent effect estimates when up to 50% of instruments are invalid
- MR-PRESSO global test for horizontal pleiotropy and outlier correction
Validation and Interpretation:
- Assess heterogeneity using Cochran's Q statistic.
- Perform "leave-one-out" analysis to determine if results are driven by single influential variants.
- For significant findings, calculate odds ratios to quantify the effect of exposure on outcome.
- For cytokines showing significant causal relationships, consider experimental validation in relevant cell models (e.g., KGN cells for POI) using Western blot and RT-PCR analyses [33].
Signaling Pathways and Genetic Networks in POI Pathogenesis
Research has identified several key signaling pathways that integrate polygenic risk in POI pathogenesis. MR studies combining genetic analyses with experimental validation have revealed that multiple risk proteins, including MCP-1/CCL2, TGFB1, ARTN, and LIFR, converge in the oncostatin M signaling pathway [33]. This pathway appears to play a central role in ovarian function and the development of POI. Additionally, pathway analyses of age at menopause GWAS loci have highlighted significant enrichment for DNA damage response (DDR) pathways, immune function, and mitochondrial biogenesis [28]. Nearly two-thirds of the genetic loci associated with age at natural menopause are involved in DDR pathways, suggesting that mechanisms maintaining genomic integrity are crucial for ovarian aging [28].
The shared genetics between age at menopause and POI further support the concept that reproductive aging may be part of systemic aging, with accumulation of DNA damage serving as a major driver [28]. Genes involved in hypothalamic-pituitary function, including FSHB, have also been identified in menopause GWAS, indicating a neuro-endocrine component to ovarian aging [28]. The enrichment of DDR genes in both natural menopause and POI suggests that these conditions exist on a continuum, with women with POI carrying more menopause-lowering variants and representing the extreme of the trait [28].
Diagram 1: Integrated Genetic and Signaling Pathways in POI Pathogenesis. This diagram illustrates how polygenic risk factors influence POI through multiple biological pathways, including DNA damage response, immune regulation, neuroendocrine function, and mitochondrial processes. Inflammatory cytokines identified through Mendelian Randomization studies modulate the immune regulation pathway.
Table 3: Essential Research Reagents and Resources for Polygenic POI Research
| Resource Category | Specific Examples | Function and Application |
|---|---|---|
| GWAS Summary Statistics | FinnGen (R10), GWAS Catalog, UK Biobank | Provide genetic association data for PRS calculation and MR instrument selection [33] [30] |
| Genotyping Arrays | Illumina Global Screening Array, UK Biobank Axiom Array | Generate genome-wide genotype data for cohort studies and PRS calculation [30] |
| LD Reference Panels | 1000 Genomes Project, HapMap | Provide linkage disequilibrium information for PRS calculation and SNP clumping [30] |
| Cell Models | KGN human granulosa-like tumor cell line | In vitro modeling of POI mechanisms and experimental validation [33] |
| Analysis Software/Packages | PRS-CS, TwoSampleMR (R package), LDpred2 | Perform PRS calculation, MR analysis, and genetic risk prediction [26] [30] |
| Experimental Validation Tools | Western blot reagents, RT-PCR systems, specific antibodies (MCP-1, LIF-R, TGF-β1, etc.) | Validate protein and gene expression changes identified through genetic studies [33] |
The integration of polygenic risk assessment and Mendelian randomization approaches has fundamentally advanced our understanding of POI pathogenesis beyond monogenic causes. Through the application of PRS, researchers can now quantify the cumulative impact of numerous genetic variants on POI risk, enabling improved risk prediction, particularly for early-onset cases. Meanwhile, MR studies have identified specific inflammatory cytokines that play causal roles in POI, revealing potential therapeutic targets and supporting the development of immunomodulatory interventions.
The convergence of findings from GWAS, PRS, and MR analyses highlights the importance of DNA damage response pathways, immune regulation, and mitochondrial function in ovarian aging and POI. These insights not only enhance our fundamental understanding of reproductive aging but also pave the way for novel diagnostic and therapeutic strategies. As genetic databases continue to expand and analytical methods become more sophisticated, the integration of polygenic risk assessment into clinical practice holds promise for early identification of at-risk individuals and personalized interventions for Premature Ovarian Insufficiency.
MR in Action: From Gene Discovery to Target Prioritization
Mendelian Randomization (MR) has emerged as a powerful epidemiological tool for investigating the causal relationships between modifiable risk factors and complex diseases, including Primary Ovarian Insufficiency (POI). By leveraging genetic variants as instrumental variables (IVs), MR can provide evidence for causal inference while minimizing confounding biases and reverse causation that often plague observational studies [31]. In the context of POI research—a condition characterized by the loss of ovarian function before age 40 affecting approximately 3.7% of women globally—MR offers a promising approach to identify genuine risk factors and potential therapeutic targets [33] [34]. The validity of any MR study hinges on fulfilling three core assumptions regarding the genetic instruments used: relevance, independence, and exclusion restriction. This document provides a detailed framework for applying these assumptions specifically within POI causal gene research, complete with experimental protocols and analytical workflows.
The Three Core MR Assumptions: Theoretical Framework and Application to POI
Assumption 1: Relevance
The relevance assumption states that the genetic instrumental variables must be robustly associated with the exposure of interest [35]. In practice, this means that single nucleotide polymorphisms (SNPs) selected as instruments must exhibit genome-wide significant associations with the exposure (e.g., inflammatory proteins, dietary factors, or gut microbiota) in prior genome-wide association studies (GWAS).
Application in POI Research: For POI studies, researchers commonly select SNPs associated with potential exposures at a significance threshold of ( P < 5 × 10^{-8} ) and ensure their strength using the F-statistic [33] [36]. For instance, in investigating inflammatory proteins as PO risk factors, Zhao et al. identified 91 inflammation-related proteins from 14,824 European participants using the Olink Target Inflammation panel [33]. Similarly, for dietary exposures, a slightly relaxed threshold (( P < 5 × 10^{-6} )) may be applied when fewer significant SNPs are available [36].
Table 1: Statistical Standards for Upholding the Relevance Assumption in POI MR Studies
| Parameter | Standard Threshold | POI-Specific Application | Key Considerations |
|---|---|---|---|
| SNP Significance | ( P < 5 × 10^{-8} ) | Applied in inflammatory protein [33] and gut microbiota studies [37] | Ensure sufficient sample size in exposure GWAS |
| F-statistic | > 10 | Calculated as ( F = \frac{R² × (N-2)}{1-R²} ) [36] | Values < 10 indicate weak instrument bias |
| LD Clustering | R² < 0.001, distance = 10,000 kb | Standard across POI MR studies [33] [34] | Ensures independence of instruments |
| Minor Allele Frequency | > 1% | Commonly applied in FinnGen and UK Biobank data | Balances instrument strength with population representativeness |
Assumption 2: Independence
The independence assumption requires that the genetic instruments must not be associated with any confounding factors that could influence the exposure-outcome relationship [31]. This assumption is grounded in Mendel's laws of inheritance, which state that genetic alleles are randomly assigned at conception, making them generally independent of environmental and lifestyle factors.
Application in POI Research: In POI studies, particular attention must be paid to confounders such as age, hormonal status, autoimmune conditions, and prior medical treatments. For example, when investigating the causal effect of gut microbiota on POI, careful consideration must be given to factors like diet, antibiotic use, and gastrointestinal disorders that could influence both microbiota composition and ovarian function [37]. The independence assumption can be evaluated using statistical methods such as MR-Egger regression and MR-PRESSO, which test for horizontal pleiotropy [33] [36].
Assumption 3: Exclusion Restriction
The exclusion restriction assumption stipulates that the genetic instruments must affect the outcome only through the exposure of interest and not via alternative biological pathways [31]. This is the most challenging assumption to verify empirically, as it requires demonstrating the absence of pleiotropic effects.
Application in POI Research: In the context of POI, violations of the exclusion restriction might occur if a genetic variant influences POI risk through multiple biological pathways. For example, a variant associated with inflammatory proteins might also affect ovarian function through direct actions on folliculogenesis rather than solely through the inflammatory pathway [33]. Sensitivity analyses are crucial for detecting such violations, including MR-Egger regression, weighted median, and mode-based estimates [33] [34].
Experimental Protocol for MR Analysis in POI Research
Stage 1: Instrument Selection and Validation
Procedure:
- Identify Exposure-Associated SNPs: Extract SNPs significantly associated with your exposure of interest (e.g., inflammatory proteins, dietary factors, gut microbiota) from relevant GWAS catalogs or previous large-scale studies.
- Clump SNPs: Perform linkage disequilibrium (LD) clumping using a reference panel (e.g., 1000 Genomes European population) with parameters R² < 0.001 and a window size of 10,000 kb to ensure independence of instruments.
- Calculate F-statistics: Compute F-statistics for each SNP using the formula: ( F = \frac{R² × (N-2)}{1-R²} ), where R² is the proportion of variance explained by the SNP and N is the sample size. Remove SNPs with F-statistics < 10 to avoid weak instrument bias.
- Harmonize Effects: Align exposure and outcome datasets so that the same effect allele is recorded in both, and remove palindromic SNPs with intermediate allele frequencies.
Table 2: Data Sources for POI MR Studies
| Data Type | Source | Sample Size | Population | POI Application |
|---|---|---|---|---|
| POI Outcome | FinnGen Consortium | 424 cases, 118,796 controls (R8) [33] | Finnish females | Primary outcome in multiple studies |
| Inflammatory Proteins | Olink Target Inflammation | 14,824 participants [33] | European | Identified causal roles of CXCL10, CX3CL1, IL-18R1 |
| Dietary Preferences | UK Biobank | 83 dietary traits [36] | European | Found dairy products increase POI risk |
| Gut Microbiota | MiBioGen Consortium | 13,266 participants [37] | Multi-ethnic | Identified protective and detrimental genera |
| Metabolites | GWAS Catalog | 50,000 participants [34] | European | Identified sphinganine-1-phosphate etc. |
Stage 2: MR Analysis Implementation
Primary Analysis:
- Employ the Inverse-Variance Weighted (IVW) method as your primary analysis, which provides a consistent causal estimate when all genetic variants are valid instruments [33] [34].
Supplementary Analyses:
- Conduct MR-Egger regression to test for directional pleiotropy, where a significant intercept indicates potential violation of the exclusion restriction assumption.
- Apply the weighted median method, which provides consistent estimates if at least 50% of the weight comes from valid instruments.
- Use the weighted mode-based approach, which remains valid when the largest number of similar causal estimates comes from valid instruments.
Sensitivity Analyses:
- Perform Cochran's Q test to assess heterogeneity among variant-specific estimates, with P < 0.05 indicating significant heterogeneity.
- Conduct MR-PRESSO global test to identify horizontal pleiotropy and correct for it by removing outliers.
- Implement leave-one-out analysis to determine if causal estimates are driven by single influential SNPs.
Stage 3: Validation and Interpretation
Validation Procedures:
- Steiger Test: Verify the direction of causality using the MR Steiger test to ensure that exposure genuinely precedes outcome [37].
- Multivariable MR: When multiple related exposures are identified, perform multivariable MR to assess their independent effects [38].
- Replication: Validate significant findings in independent datasets where available to enhance robustness.
- Biological Plausibility: Interpret significant MR results in the context of existing biological knowledge about POI pathophysiology.
Visualization of MR Workflow and Analytical Framework
MR Analytical Workflow
Table 3: Essential Research Reagents and Computational Tools for POI MR Studies
| Resource Type | Specific Tool/Database | Application in POI MR | Key Features |
|---|---|---|---|
| Statistical Software | R package "TwoSampleMR" [33] [36] | Primary MR analysis | Comprehensive suite for two-sample MR |
| GWAS Database | FinnGen Consortium (R8/R11) [33] [34] | POI outcome data | 424-542 cases, 118,796-241,998 controls |
| Protein GWAS | Olink Target Inflammation [33] | Inflammation exposure | 91 inflammation-related proteins |
| Microbiome GWAS | MiBioGen Consortium [37] | Gut microbiota exposure | 211 microbial taxa, 13,266 individuals |
| Pleiotropy Detection | MR-PRESSO [36] [37] | Exclusion restriction validation | Identifies and corrects for horizontal pleiotropy |
| Sensitivity Analysis | MR-Egger regression [33] [34] | Independence assumption testing | Evaluates directional pleiotropy |
| Data Harmonization | LDlinkR [36] | Instrument preparation | Linkage disequilibrium reference |
Case Study: Applying MR Assumptions in POI Inflammation Research
A recent MR study investigating inflammatory proteins in POI provides an exemplary model for applying the three core assumptions [33]. The researchers began by selecting instruments for 91 inflammation-related proteins from GWAS data involving 14,824 European participants, ensuring relevance through stringent significance thresholds (( P < 5 × 10^{-8} )) and F-statistics > 10. To uphold the independence assumption, they conducted comprehensive sensitivity analyses including MR-Egger intercept tests and MR-PRESSO global tests. For the exclusion restriction assumption, they employed multiple complementary methods (weighted median, mode) and validation experiments in POI cell models.
This approach identified CXCL10 and CX3CL1 as protective against POI, while IL-18R1, IL-18, MCP-1, and CCL28 increased POI risk. Subsequent gene-drug analysis identified CCL2 and TGFB1 as potential therapeutic targets, with genistein and melatonin prioritized as potential treatments [33]. This study demonstrates how rigorous application of MR assumptions can yield biologically plausible and clinically relevant insights into POI pathogenesis.
The three core MR assumptions—relevance, independence, and exclusion restriction—provide the foundational framework for valid causal inference in POI research. As MR methodologies continue to evolve and larger GWAS datasets become available, adherence to these assumptions will remain paramount for generating reliable evidence regarding the causal determinants of POI. The protocols and guidelines outlined in this document provide a roadmap for researchers to implement robust MR studies that can ultimately contribute to improved prevention, diagnosis, and treatment strategies for this clinically significant condition.
The discovery of causal genes for complex diseases like Primary Ovarian Insufficiency (POI) remains a significant challenge in genomics research. Mendelian randomization (MR) has emerged as a powerful statistical framework that uses genetic variants as instrumental variables to infer causal relationships between molecular traits and disease outcomes [33]. By integrating multi-omics quantitative trait loci data, including expression QTLs (eQTLs) and protein QTLs (pQTLs), researchers can bridge the gap between statistical associations and biological mechanisms in POI pathogenesis [39] [40].
This protocol details the application of multi-omics data integration within an MR framework, specifically leveraging resources from the Genotype-Tissue Expression (GTEx) project and the eQTLGen Consortium to identify and validate causal genes for POI. The integration of eQTL and pQTL data enables researchers to move beyond genetic associations to understand the functional consequences of genetic variants across molecular layers [41] [40].
Background
Primary Ovarian Insufficiency and the Need for Causal Gene Discovery
POI is a clinically heterogeneous condition characterized by the loss of ovarian function before age 40, affecting approximately 3.7% of women globally [34]. The condition presents with menstrual disturbances, elevated gonadotropins, and infertility, often accompanied by increased risks of osteoporosis and cardiovascular disease [42]. Current treatments are primarily symptomatic, focusing on hormone replacement and fertility preservation, with limited efficacy due to incomplete understanding of POI pathogenesis [33].
Traditional genome-wide association studies (GWAS) have identified numerous loci associated with POI risk, but most reside in non-coding regions with unclear functional significance [34]. This limitation underscores the need for approaches that can prioritize causal genes and elucidate their mechanisms of action.
eQTL and pQTL Fundamentals
Expression quantitative trait loci (eQTLs) represent genetic variants that influence gene expression levels, while protein quantitative trait loci (pQTLs) affect protein abundance [41]. These molecular QTLs serve as crucial functional interpreters of GWAS signals, helping to identify which genetic associations likely operate through regulation of specific genes or proteins.
Large-scale consortia have generated comprehensive eQTL and pQTL resources:
- The GTEx project provides eQTL data across 54 non-diseased tissues from nearly 1,000 donors, enabling tissue-specific regulatory inference [41] [40].
- The eQTLGen Consortium offers eQTL data from 31,684 individuals, primarily from blood tissue [39] [34].
- pQTL datasets from studies like deCODE (35,559 Icelandic individuals) provide plasma protein abundance data [39] [40].
Table 1: Major QTL Data Sources for POI Research
| Resource | Data Type | Sample Size | Tissues/Cell Types | Primary Applications |
|---|---|---|---|---|
| GTEx v8 | eQTL | ~1,000 donors | 54 tissues including ovary | Tissue-specific regulatory mechanisms |
| eQTLGen Consortium | eQTL | 31,684 individuals | Whole blood | Large-scale cis and trans-eQTL discovery |
| deCODE Genetics | pQTL | 35,559 individuals | Plasma | Protein-disease causal inference |
| OneK1k Project | sc-eQTL | 982 donors | Peripheral blood mononuclear cells | Cell-type-specific regulation |
Mendelian Randomization Framework for Causal Inference
MR relies on three core assumptions for valid causal inference:
- Relevance: Genetic instruments must be strongly associated with the exposure (e.g., gene expression)
- Exchangeability: Instruments must not be associated with confounders
- Exclusion restriction: Instruments affect outcome only through the exposure [33] [42]
In multi-omics MR, these assumptions are extended to integrate evidence across molecular layers, strengthening causal inference when consistent effects are observed across omics levels [39] [40].
GTEx Data Access and Quality Control
The GTEx portal (https://gtexportal.org/) provides comprehensive eQTL data from multiple tissues. For POI research, ovarian tissue data is particularly relevant, though sample sizes may be limited. When ovary data is insufficient, researchers can utilize cross-tissue resources or meta-analysis approaches.
Processing steps:
- Download normalized transcript per million (TPM) values and covariate files
- Apply quality filters: RIN > 6.0, genotyping rate > 0.95
- Account for hidden confounders using PEER factors
- Transform data using rank-based inverse normal transformation
eQTLGen Consortium Data
The eQTLGen Consortium (https://eqtlgen.org/) provides cis- and trans-eQTLs from blood tissue, with a sample size of 31,684 individuals. While blood may not be the most relevant tissue for POI, its large sample size provides excellent statistical power for initial discovery.
Processing steps:
- Download preprocessed summary statistics
- Apply significance threshold (P < 5×10⁻⁸ for cis-eQTLs)
- Extract lead independent SNPs using LD clumping (r² < 0.1)
pQTL data can be obtained from several sources:
- deCODE study: 35,559 Icelandic individuals, 4,907 plasma proteins [39] [40]
- UK Biobank Pharma Proteomics Project: 54,306 participants, 2,924 plasma proteins [34]
Processing steps:
- Apply significance thresholds (P < 5×10⁻⁸ for cis-pQTLs)
- Account for technical covariates (batch effects, sample handling)
- Normalize protein levels using appropriate methods (e.g., quantile normalization)
POI GWAS Data
FinnGen Consortium (https://r11.finngen.fi/) provides POI GWAS summary statistics (542 cases, 241,998 controls) [34]. Alternative sources include the R10 release with 424 cases and 118,796 controls [33].
Processing steps:
- Apply standard QC filters: INFO > 0.8, MAF > 0.01
- Remove strand-ambiguous and palindromic SNPs
- Allele harmonization across datasets
Table 2: Instrumental Variable Selection Criteria by Data Type
| Data Type | P-value Threshold | LD Clumping Parameters | F-statistic Calculation | Minimum F-statistic |
|---|---|---|---|---|
| cis-eQTL | P < 5×10⁻⁸ | r² < 0.1, window=10,000 kb | F = (R²/K) × ((n - k - 1)/(1 - R²)) | F > 10 |
| cis-pQTL | P < 5×10⁻⁸ | r² < 0.1, window=10,000 kb | F = (R²/K) × ((n - k - 1)/(1 - R²)) | F > 10 |
| trans-eQTL | P < 1×10⁻⁵ | r² < 0.001, window=10,000 kb | F = (R²/K) × ((n - k - 1)/(1 - R²)) | F > 10 |
| POI GWAS | P < 5×10⁻⁸ | r² < 0.001, window=10,000 kb | N/A | N/A |
Integrative Analysis Workflow
The following diagram illustrates the comprehensive workflow for integrating multi-omics data in Mendelian randomization studies of POI:
Two-Sample Mendelian Randomization
The core analysis employs two-sample MR using the TwoSampleMR R package (v0.5.7) [39]. This approach uses genetic instruments from eQTL/pQTL studies to estimate their causal effect on POI risk.
Primary method: Inverse-variance weighted (IVW) regression provides the main causal estimate under the assumption that all instruments are valid [33] [34].
Supplementary methods:
- MR-Egger: Accounts for directional pleiotropy through intercept testing
- Weighted median: Consistent estimate when >50% of weight comes from valid instruments
- Weighted mode: Clusters instruments based on causal estimates
Implementation code:
SMR analysis integrates eQTL and GWAS data to test whether genetic effects on POI are mediated through gene expression [34] [40]. The HEIDI test distinguishes pleiotropy from linkage by testing heterogeneity in causal effect estimates across multiple SNPs in a locus.
Implementation:
Interpretation:
- Significant SMR p-value (P < 2.63×10⁻⁶ after Bonferroni correction) suggests causal association
- HEIDI p-value > 0.01 indicates consistent effect across SNPs (supports causality)
Colocalization Analysis
Colocalization analysis determines whether eQTL/pQTL and POI GWAS signals share a common causal variant using the coloc R package (v5.2.3) [40].
Hypotheses tested:
- H0: No association with either trait
- H1: Association with eQTL/pQTL only
- H2: Association with POI only
- H3: Association with both, different causal variants
- H4: Association with both, shared causal variant
Implementation:
A posterior probability for H4 (PP.H4) > 0.8 provides strong evidence for colocalization [40].
Sensitivity Analysis and Validation
Sensitivity Analyses
Cochran's Q statistic assesses heterogeneity across instrumental variables, with P < 0.05 indicating significant heterogeneity [33].
MR-Egger intercept test evaluates directional pleiotropy (P < 0.05 suggests significant pleiotropy) [39] [42].
MR-PRESSO identifies and corrects for outliers in the MR analysis [42].
Leave-one-out analysis examines if causal estimates are driven by single influential SNPs.
Cross-Tissue and Cross-Omics Consistency
Compare effect directions and magnitudes across:
- Multiple tissues (ovary, blood, brain)
- Molecular levels (eQTL vs pQTL)
- Independent datasets
Consistent effects across tissues and omics layers strengthen causal inference. For example, in a recent POI study, MCP-1/CCL2 and TGFB1 showed consistent evidence across proteomic and genetic analyses [33].
Case Study: Application to POI Research
A recent MR study investigating inflammatory proteins in POI identified several potential causal factors [33]:
Table 3: Exemplar Causal Findings in POI from Multi-omics MR
| Gene/Protein | Omics Evidence | Direction of Effect | OR (95% CI) | P-value | Supporting Evidence |
|---|---|---|---|---|---|
| CXCL10 | pQTL | Protective | 0.87 (0.82-0.93) | 4.2×10⁻⁵ | Wald ratio, IVW |
| CX3CL1 | pQTL | Protective | 0.92 (0.88-0.96) | 3.8×10⁻⁴ | IVW |
| IL-18R1 | pQTL | Risk | 1.14 (1.07-1.21) | 6.5×10⁻⁵ | IVW |
| MCP-1/CCL2 | pQTL, Experimental | Risk | 1.18 (1.09-1.28) | 2.3×10⁻⁵ | IVW, Western blot |
| TGFB1 | pQTL, Experimental | Risk | 1.22 (1.11-1.34) | 1.7×10⁻⁵ | IVW, RT-PCR |
The analysis workflow for this study can be visualized as follows:
The Scientist's Toolkit
Table 4: Essential Research Reagent Solutions for Multi-omics POI Research
| Resource Type | Specific Examples | Function/Application | Access Information |
|---|---|---|---|
| QTL Databases | GTEx Portal (v8), eQTLGen, deCODE pQTL | Source of genetic instruments for MR analysis | https://gtexportal.org/, https://eqtlgen.org/ |
| GWAS Catalogs | FinnGen (R11), GWAS Catalog | POI outcome data for causal inference | https://r11.finngen.fi/, https://www.ebi.ac.uk/gwas/ |
| Software Packages | TwoSampleMR (R), SMR, COLOC | Statistical analysis of causal relationships | https://mrcieu.github.io/TwoSampleMR/ |
| Experimental Validation | KGN cell line, cyclophosphamide model | Functional validation of MR findings | Commercial vendors (e.g., iCell Bioscience) |
| Pathway Databases | KEGG, MSigDB, miEAA | Biological interpretation of findings | https://www.genome.jp/kegg/, https://www.gsea-msigdb.org/ |
| Drug Repurposing | DGIdb database | Identification of potential therapeutic compounds | https://www.dgidb.org/ |
Troubleshooting and Technical Considerations
Common Analytical Challenges
Weak instrument bias: Ensure all SNPs have F-statistics > 10 to minimize bias [39]. If instruments are weak, consider:
- Relaxing significance thresholds (P < 1×10⁻⁵)
- Using Bayesian methods that account for instrument strength
Horizontal pleiotropy: When MR-Egger intercept test indicates pleiotropy (P < 0.05):
- Apply MR-PRESSO to remove outliers
- Use weighted median or mode methods
- Conduct sensitivity analyses excluding pleiotropic SNPs
Sample overlap: Use independent samples for exposure and outcome when possible. If overlap exists, apply correction methods.
LD contamination: Use stringent LD clumping (r² < 0.001) and verify results with HEIDI test.
Tissue Relevance Considerations
For POI research, ovarian tissue is most relevant but sample sizes are limited. Consider:
- Using blood eQTLs as proxies with large sample sizes
- Incorporating single-cell eQTL data from relevant cell types
- Testing consistency across available tissues
This protocol outlines a comprehensive framework for integrating eQTL and pQTL data from GTEx and eQTLGen to identify causal genes for POI using Mendelian randomization. The multi-omics approach strengthens causal inference by providing consistent evidence across molecular levels, from genetic variation to gene expression to protein function.
The application of these methods has already yielded promising candidates for POI, including inflammatory proteins like MCP-1/CCL2 and TGFB1, which converge on the oncostatin M signaling pathway [33]. Future directions include incorporating single-cell QTL data, expanding to non-European populations, and integrating additional omics layers such as methylation and metabolomics to further elucidate the molecular architecture of POI.
Mendelian randomization (MR) has emerged as a powerful statistical technique in epidemiological research, using genetic variants as instrumental variables (IVs) to infer causal relationships between modifiable exposures and health outcomes [43]. This approach is particularly valuable for investigating the etiology of complex diseases such as premature ovarian insufficiency (POI), where randomized controlled trials are often impractical or unethical [5]. The method leverages the natural randomization of genetic alleles at conception, which reduces confounding and eliminates reverse causation concerns inherent in traditional observational studies [44].
Within the specific context of POI research, MR offers a promising avenue to identify causal risk factors and potential therapeutic targets. POI affects approximately 1-3% of women under 40 and represents a significant clinical challenge in reproductive medicine [5]. Recent studies have begun to apply MR frameworks to identify noninvasive biomarkers and causal metabolites for POI, demonstrating the methodology's practical utility in this field [5] [45]. This protocol provides a comprehensive, step-by-step workflow for implementing MR analysis specifically tailored to POI research, from data acquisition through causal estimation and sensitivity analysis.
Materials
Research Reagent Solutions
Table 1: Essential materials and computational tools for Mendelian randomization analysis
| Item | Function | Example Sources/Platforms |
|---|---|---|
| GWAS Summary Data | Source data for exposure and outcome variables | FinnGen database, IEU OpenGWAS database, GWAS Catalog [46] [5] |
| TwoSampleMR R Package | Data management, harmonization, and statistical analysis for MR | MRCIEU GitHub repository [46] |
| ieugwasr R Package | Programmatic access to IEU OpenGWAS database | MRCIEU GitHub repository [46] |
| LD Reference Panel | Linkage disequilibrium estimation for clumping | 1000 Genomes Project [45] |
| Cis-eQTL Data | Integration of expression quantitative trait loci | eQTLGen Consortium [5] |
| Cis-pQTL Data | Integration of protein quantitative trait loci | deCODE Genetics, UK Biobank [47] [44] |
Software and System Requirements
The workflow requires R (version 4.0 or higher) and specific packages as detailed in Table 1. The TwoSampleMR package can be installed from the MRCIEU R Universe repository using the following code:
Method
Instrument Selection
The initial step involves selecting appropriate genetic instruments for the exposure variable. For POI research, this typically involves extracting single nucleotide polymorphisms (SNPs) associated with the exposure of interest (e.g., metabolites, proteins, or other risk factors) from GWAS summary statistics.
Procedure:
- Set Significance Threshold: Apply a genome-wide significance threshold (typically P < 5×10^-8 for strong instruments, though P < 1×10^-5 may be used for metabolic traits) [5] [45].
- Perform LD Clumping: Use the
clump_datafunction in TwoSampleMR with a reference panel (e.g., 1000 Genomes European population) to select independent SNPs (r^2^ < 0.001 within a 10,000 kb window) [5]. - Calculate F-statistic: Assess instrument strength using F = (β~exposure~/SE~exposure~)^2^. Retain SNPs with F > 10 to minimize weak instrument bias [45].
Outcome Data Extraction
After selecting instruments, extract their corresponding effect estimates from the outcome GWAS summary statistics (e.g., POI data).
Procedure:
- Identify Outcome Dataset: Obtain POI GWAS summary statistics from databases such as FinnGen (e.g., R11 release with 542 cases and 241,998 controls) [5].
- Extract Outcome Effects: Use the
extract_outcome_datafunction to retrieve effect estimates for the selected instruments.
Data Harmonization
Harmonization ensures that effect alleles are aligned between exposure and outcome datasets, which is crucial for valid causal estimates.
Procedure:
- Align Effect Alleles: Ensure the same effect allele is used for both exposure and outcome effects.
- Palindromic SNPs: Remove or resolve palindromic SNPs (those with A/T or G/C alleles) where the strand orientation is ambiguous.
- Effect Alignment: Use the
harmonise_datafunction to automatically harmonize effect alleles and effect sizes.
MR Analysis
Perform the primary MR analysis using multiple complementary methods to ensure robust causal inference.
Procedure:
- Primary Analysis: Apply the inverse variance weighted (IVW) method as the primary analysis when multiple valid instruments are available.
- Supplementary Analyses: Implement additional methods including MR-Egger, weighted median, weighted mode, and contamination mixture methods to assess robustness.
- Sensitivity Analysis: Conduct heterogeneity and pleiotropy tests to validate MR assumptions.
Sensitivity Analyses
Comprehensive sensitivity analyses are essential to validate MR assumptions and ensure result robustness.
Procedure:
- Pleiotropy Assessment: Use the MR-Egger intercept test to assess directional pleiotropy (P < 0.05 suggests significant pleiotropy) [5].
- Heterogeneity Testing: Calculate Cochran's Q statistic to detect heterogeneity among IV estimates (P < 0.05 indicates significant heterogeneity) [5].
- Leave-One-Out Analysis: Perform leave-one-out analysis to determine if causal estimates are driven by individual SNPs.
- MR-PRESSO: Apply MR-PRESSO to detect and correct for horizontal pleiotropic outliers.
Expected Results
Primary Causal Estimates
Table 2: Example MR results for metabolites causally associated with POI risk [45]
| Metabolite | OR (95% CI) | P-value | FDR | Method |
|---|---|---|---|---|
| Sphinganine-1-phosphate | 1.52 (1.28-1.80) | 2.1×10^-6^ | 0.03 | IVW |
| X-23636 | 0.65 (0.52-0.81) | 1.8×10^-4^ | 0.04 | IVW |
| 4-methyl-2-oxopentanoate | 1.48 (1.22-1.79) | 6.3×10^-5^ | 0.04 | IVW |
| Faecalibacterium abundance | 0.61 (0.45-0.82) | 0.001 | 0.04 | IVW |
Sensitivity Analysis Results
Table 3: Expected sensitivity analysis outputs for POI MR analysis
| Test | Statistic | Interpretation |
|---|---|---|
| MR-Egger Intercept | P > 0.05 | No significant directional pleiotropy |
| Cochran's Q (IVW) | P > 0.05 | No significant heterogeneity |
| MR-PRESSO Global Test | P > 0.05 | No significant horizontal pleiotropy |
| F-statistic | > 10 | Adequate instrument strength |
Troubleshooting
Common Issues and Solutions
- Weak Instrument Bias: If F-statistics < 10, consider using a less stringent significance threshold (P < 1×10^-5) for instrument selection or applying methods robust to weak instruments such as MR-RAPS [45].
- Horizontal Pleiotropy: If MR-Egger intercept indicates significant pleiotropy (P < 0.05), consider using robust methods such as weighted median, MR-PRESSO, or MRMix that account for pleiotropic effects [48] [43].
- Sample Overlap: In cases of sample overlap between exposure and outcome datasets, apply methods that correct for bias due to overlap, such as the MRCI framework [48].
- Binary Outcomes: For binary outcomes like POI, ensure causal estimates are presented as odds ratios with 95% confidence intervals, and consider using the delta method for accurate standard error calculation [43].
Workflow Visualization
Figure 1: Mendelian randomization workflow from GWAS summary data to causal estimate interpretation
Time Requirements
A complete MR analysis following this protocol typically requires 2-4 hours of computational time, depending on dataset size and complexity. Data preparation and harmonization constitute approximately 30% of the time, primary MR analysis 20%, and sensitivity analyses the remaining 50%. These estimates assume standard computing resources (8 GB RAM, 4-core processor) and moderately sized GWAS datasets (< 1 million SNPs).
Primary ovarian insufficiency (POI) is a clinically significant disorder characterized by the loss of ovarian function before the age of 40, affecting approximately 3.7% of women globally and leading to substantial impacts on fertility, bone health, cardiovascular function, and overall quality of life [49] [50] [5]. The etiology of POI remains incompletely understood, which has hindered the development of targeted and effective therapeutic strategies. Current management primarily relies on hormone replacement therapy (HRT), which addresses symptoms but does not restore ovarian function or fertility [51] [50]. A significant pathological feature is that many women with POI retain dormant primordial follicles in their ovaries, suggesting that therapeutic interventions aimed at "reawakening" these follicles could restore ovarian function [49].
Mendelian randomization (MR) has emerged as a powerful epidemiological method that uses genetic variants as instrumental variables to infer causal relationships between modifiable exposures and disease outcomes. By leveraging the random allocation of genetic alleles at conception, MR minimizes confounding and reverse causation biases that often plague traditional observational studies [52] [53]. In the context of POI, MR analysis is particularly valuable for identifying causal genetic factors and prioritizing potential therapeutic targets for further investigation.
This case study details the application of an integrated genomic approach, combining genome-wide association studies (GWAS) with expression quantitative trait loci (eQTL) data and MR methodology, to identify and validate FANCE and RAB2A as promising therapeutic targets for POI treatment.
Results
Identification of Potential Causal Genes for POI
The initial genome-wide Mendelian randomization analysis investigated the association between 431 genes with available index cis-eQTL signals and POI risk. After rigorous statistical correction and sensitivity analyses to exclude pleiotropic effects, four genes demonstrated statistically significant associations with reduced risk of POI [49].
Table 1: Genes Significantly Associated with POI Risk via Mendelian Randomization
| Gene | eQTL Data Source | Odds Ratio (95% CI) | P-value | Bonferroni-corrected P |
|---|---|---|---|---|
| HM13 | Whole Blood (GTEx V8) | 0.76 (0.66–0.88) | 0.0003 | 0.046 |
| FANCE | Ovary (GTEx V8) | 0.82 (0.72–0.93) | 0.0003 | 0.018 |
| RAB2A | eQTLGen Consortium | 0.73 (0.62–0.86) | 0.0001 | 0.036 |
| MLLT10 | eQTLGen Consortium | 0.74 (0.64–0.86) | 0.00008 | 0.022 |
The results indicated that increased expression of these genes is causally associated with a protective effect against POI, with odds ratios significantly below 1.0 [49] [54].
Colocalization Analysis Validates FANCE and RAB2A
To distinguish true causal relationships from mere linkage disequilibrium, researchers performed Bayesian colocalization analysis. This analysis calculates posterior probabilities for different hypotheses regarding shared causal variants between gene expression and POI risk [49].
Table 2: Colocalization Analysis Results for Candidate POI Genes
| Gene | PP.H4 (Same Causal Variant) | Colocalization Support |
|---|---|---|
| FANCE | 0.86 | Strong |
| RAB2A | 0.91 | Strong |
| HM13 | 0.78 | Moderate |
| MLLT10 | 0.01 | Weak |
The analysis provided strong colocalization evidence for FANCE and RAB2A, with posterior probabilities (PP.H4) of 0.86 and 0.91, respectively. This indicates a high probability that the same underlying genetic variant influences both the expression of these genes and POI risk, strengthening their candidacy as therapeutic targets [49] [54].
Biological Rationale and Druggability Assessment
A comprehensive assessment of the biological functions and druggability potential of the identified genes revealed compelling rationales for FANCE and RAB2A:
FANCE: This gene encodes a core component of the Fanconi anemia (FA) DNA repair pathway, which is crucial for the repair of DNA interstrand crosslinks. Proper DNA repair is essential for maintaining oocyte genomic integrity and preventing follicle depletion. Its involvement in this fundamental cellular process makes it a compelling target for therapeutic modulation [49].
RAB2A: This gene encodes a member of the RAS oncogene family involved in regulating autophagy and vesicular trafficking. Autophagy plays a critical role in folliculogenesis and oocyte development. Dysregulation of these processes could contribute to premature follicle loss, positioning RAB2A as a key regulator of ovarian homeostasis [49].
Both FANCE and RAB2A were classified as promising druggable candidates based on their biological functions, with FANCE involved in DNA repair and RAB2A in autophagy regulation—both processes amenable to pharmacological intervention [49].
Discussion
The identification of FANCE and RAB2A as potential therapeutic targets for POI represents a significant advancement in the field of reproductive medicine. The strength of this case study lies in the rigorous application of Mendelian randomization framework, which provides robust evidence for a causal relationship between these genes and POI risk, moving beyond mere association [49] [52].
The biological plausibility of both targets is well-supported by their roles in critical cellular processes. FANCE's involvement in DNA repair is particularly relevant given the sensitivity of oocytes to DNA damage accumulation over time. The RAB2A-autophagy axis represents a novel pathway for therapeutic exploration in ovarian biology, potentially offering new mechanisms to modulate follicular activation and survival [49].
From a drug development perspective, several strategic considerations emerge:
Target Modulation Strategy: For both FANCE and RAB2A, the therapeutic goal would be to enhance their expression or activity, given their protective effect against POI. This presents a different challenge compared to traditional inhibitor development.
Alternative Inflammatory Targets: Parallel research on inflammation-related proteins in POI has identified additional potential targets, including MCP-1/CCL2 and TGFB1, with genistein and melatonin prioritized as potential therapeutic compounds [51].
Pathway-Based Approaches: Enrichment analyses of POI-related genes and miRNAs have highlighted potential involvement of pathways such as glutathione metabolism and the PI3K pathway, offering alternative intervention points [5].
This study also demonstrates the power of integrating multi-omics data (genomics, transcriptomics) through MR methodology to elucidate the genetic architecture of complex disorders like POI. This approach can be extended to incorporate additional omics layers, including proteomics and metabolomics, to further refine our understanding of POI pathophysiology [5].
Further validation in in vitro and in vivo models is necessary to confirm the therapeutic potential of modulating FANCE and RAB2A before clinical translation. Additionally, exploration of these targets may have implications beyond POI, potentially benefiting women with other forms of ovarian dysfunction or age-related fertility decline.
Methods
This investigation employed a multi-tiered analytical approach combining GWAS summary data with expression quantitative trait loci (eQTL) information through Mendelian randomization and colocalization techniques.
Mendelian Randomization and Colocalization Protocol
Purpose: To test for causal effects of gene expression on POI risk by integrating eQTL and GWAS data [49].
Procedure:
- Software Implementation: Perform SMR analysis using SMR software tool (version 1.3.1)
- Heterogeneity Testing: Conduct HEIDI test to address potential pleiotropy
- Apply threshold of PHEIDI < 0.05 for exclusion of genes with significant pleiotropy
- Two-sample MR: Perform additional validation using Wald ratio and delta method
- Calculate odds ratios (OR) and 95% confidence intervals (CI)
- Statistical Significance: Apply Bonferroni correction with threshold of P < 0.05
Colocalization Analysis Protocol
Purpose: To distinguish causal associations from linkage disequilibrium by assessing whether gene expression and POI risk share the same causal genetic variant [49].
Procedure:
- Software Implementation: Utilize coloc R package with Bayesian approach
- Hypothesis Testing: Calculate posterior probabilities for five hypotheses:
- PP.H0: No association with either trait
- PP.H1: Association with gene expression only
- PP.H2: Association with POI only
- PP.H3: Association with both traits but different causal variants
- PP.H4: Association with both traits with same causal variant
- Parameter Settings: Apply default priors (p1 = 1×10−4, p2 = 1×10−4, p12 = 1×10−5)
- Interpretation Criteria: Restrict analysis to genes with PP.H3 + PP.H4 ≥ 0.8 for adequate power
Druggability Assessment Protocol
Purpose: To evaluate the potential of identified genes as therapeutic targets [49].
Procedure:
- Database Queries: Search multiple biological and pharmacological databases:
- Online Mendelian Inheritance in Man (OMIM)
- DrugBank database
- Drug-Gene Interaction database (DGIdb)
- Therapeutic Target Database (TTD)
- Evaluation Criteria: Assess targets based on:
- Approval status for marketing or involvement in clinical trials
- Preclinical development stage
- General druggability based on protein characteristics and biological function
- Expert Review: Classify targets as promising even without database documentation if supported by biological rationale
The Scientist's Toolkit
Table 3: Essential Research Reagents and Resources for POI Therapeutic Target Identification
| Category | Specific Resource | Function/Application | Source/Reference |
|---|---|---|---|
| GWAS Data | FinnGen R11 Dataset | Provides summary statistics for POI cases and controls | [49] |
| eQTL Data | GTEx V8 (Ovary) | Tissue-specific gene expression regulation data | [49] |
| eQTL Data | eQTLGen Consortium | Large-scale blood eQTL reference | [49] |
| Analysis Tools | SMR Software (v1.3.1) | Integrates eQTL and GWAS data for causal inference | [49] |
| Analysis Tools | coloc R Package | Bayesian colocalization analysis | [49] |
| Databases | OMIM, DrugBank, DGIdb, TTD | Druggability assessment and target validation | [49] |
| Cell Models | KGN Human Granulosa Cells | In vitro modeling of POI mechanisms | [51] |
Visualizing Biological Pathways
Phenome-Wide Mendelian Randomization (PheWAS-MR) represents a paradigm shift in causal inference research, moving beyond the traditional single-exposure-single-outcome framework to systematically evaluate thousands of exposure-outcome relationships simultaneously. In the context of Premature Ovarian Insufficiency (POI) research, this hypothesis-free approach enables researchers to uncover novel risk factors, biomarkers, and therapeutic targets without prior assumptions about disease etiology. The core strength of PheWAS-MR lies in its ability to detect pleiotropic effects—whereby genetic variants influence multiple traits—thereby providing a more comprehensive understanding of the complex biological networks underlying POI pathogenesis.
The methodology integrates two powerful epidemiological approaches: Mendelian randomization, which uses genetic variants as instrumental variables to infer causal relationships, and phenome-wide association studies, which systematically test associations across a wide range of phenotypes. When applied to POI, a condition affecting approximately 3.7% of women globally and characterized by loss of ovarian function before age 40, PheWAS-MR offers particular promise for addressing critical challenges in disease management [34]. Specifically, it can identify non-invasive warning markers for early detection and illuminate potential pathways for therapeutic intervention in a condition that currently lacks effective treatments [34].
Theoretical Foundations and Analytical Framework
Core Principles and Assumptions
PheWAS-MR rests on three fundamental assumptions that must be satisfied for valid causal inference. First, genetic instruments must exhibit robust associations with the exposure traits of interest. Second, these instruments must be independent of confounders affecting the exposure-outcome relationship. Third, genetic variants must influence the outcome exclusively through the exposure, not via alternative biological pathways (the exclusion restriction criterion) [33] [34]. The random assortment of genetic variants at conception helps mitigate confounding and reverse causation biases that often plague conventional observational studies [55].
In POI research, particular attention must be paid to vertical pleiotropy (where a genetic variant affects multiple traits along a causal pathway) versus horizontal pleiotropy (where a variant influences multiple traits through independent pathways), as distinguishing between these is crucial for accurate biological interpretation. The PheWAS-MR framework employs several statistical approaches to address these challenges, including sensitivity analyses and robust MR methods that can detect and adjust for pleiotropic effects [56].
Study Design Considerations
Implementing a robust PheWAS-MR study for POI requires careful consideration of several design elements. Researchers must define the phenome scope, which typically encompasses thousands of traits across diverse categories including anthropometric measures, biomarkers, dietary factors, and clinical conditions. For POI applications, the FinnGen database has emerged as a valuable resource, providing summary statistics from 424 Finnish adult female POI cases and 118,796 controls [33], though sample size limitations remain a constraint that should be acknowledged.
Instrument selection represents another critical consideration. Genetic instruments are typically single nucleotide polymorphisms (SNPs) meeting genome-wide significance thresholds (P < 5×10⁻⁸) and clumped to ensure independence (linkage disequilibrium R² < 0.001) [33]. The strength of these instruments is commonly assessed using the F-statistic, with values greater than 10 indicating sufficient strength to minimize weak instrument bias [34]. For molecular traits such as protein levels, cis-acting variants (located within 500kb of the encoding gene) are often preferred due to their higher biological prior and reduced likelihood of pleiotropy [56] [57].
Table 1: Key Database Resources for POI PheWAS-MR Studies
| Database/Resource | Description | Sample Characteristics | POI-Relevant Applications |
|---|---|---|---|
| FinnGen Consortium | GWAS summary statistics for POI | 424 cases, 118,796 controls (Finnish) [33] | Primary outcome data for POI |
| Olink Target Inflammation Panel | 91 inflammation-related proteins | 14,824 European participants [33] | Inflammatory mechanisms in POI |
| eQTLGen Consortium | Expression quantitative trait loci | 31,684 individuals [34] | SMR analysis for gene expression |
| UK Biobank Proteomics | 2,904 plasma proteins | 54,306 participants [34] | Proteome-wide causal inference |
Analytical Protocols for POI-Focused PheWAS-MR
Primary MR Analysis Workflow
The foundational analytical protocol for PheWAS-MR begins with quality control of genetic instruments and proceeds through several analytical steps. For each of the thousands of exposure-outcome pairs tested, the following protocol is recommended:
Step 1: Instrument Extraction and Clumping Extract genome-wide significant SNPs (P < 5×10⁻⁸) associated with each exposure trait from relevant GWAS summary statistics. Perform LD clumping (R² < 0.001 within 10,000 kb window) to ensure instrument independence using a reference panel such as 1000 Genomes [58]. Calculate F-statistics for each instrument (F = [R²(n-2)]/[1-R²], where R² is the proportion of variance explained) and exclude variants with F < 10 to avoid weak instrument bias [33].
Step 2: Effect Size Harmonization Harmonize exposure and outcome effects to ensure all SNPs are aligned to the same effect allele. Carefully manage palindromic SNPs by comparing allele frequencies with reference data or excluding if frequencies are ambiguous.
Step 3: Primary MR Analysis Perform two-sample MR using the inverse variance weighted (IVW) method as the primary analysis for exposures with multiple instruments. For exposures with only one instrument, use the Wald ratio method. Apply random-effects IVW when heterogeneity is detected [58].
Step 4: Multiple Testing Correction Account for the massive multiple testing burden in PheWAS-MR by implementing hierarchical significance thresholds. For POI applications with ~3,000 exposures, consider: "robust" evidence (P < 1.67×10⁻⁵, Bonferroni-corrected for 3,000 tests), "probable" evidence (P < 0.001), and "suggestive" evidence (P < 0.05) [55].
Sensitivity Analysis and Validation
Robust PheWAS-MR requires extensive sensitivity analyses to validate findings and address potential violations of MR assumptions:
Step 5: Pleiotropy Assessment Apply the MR-Egger method and examine its intercept term to assess directional pleiotropy. A statistically significant MR-Egger intercept (P < 0.05) suggests the presence of unbalanced pleiotropy that may bias causal estimates [33].
Step 6: Heterogeneity Testing Calculate Cochran's Q statistic to detect heterogeneity among variant-specific causal estimates. Significant heterogeneity (P < 0.05) may indicate pleiotropy or other violations of MR assumptions [34].
Step 7: Leave-One-Out Analysis Iteratively remove each SNP and re-run MR analyses to identify influential variants that disproportionately drive causal estimates.
Step 8: Colocalization Analysis For significant associations, perform colocalization analysis to determine whether exposure and outcome share the same causal variant. A posterior probability > 80% provides strong evidence against coincidental linkage disequilibrium [56]. Methods such as PWCoCo (Pairwise Conditional and Colocalization) can handle regions with multiple independent signals [56].
Step 9: Independent Validation Replicate significant findings in independent datasets where possible. For POI, the FinnGen cohort provides a valuable validation resource [58].
Advanced cis-MR Methods for Drug Target Discovery
For investigating specific molecular traits such as protein biomarkers in POI, cis-MR methods focusing on genetic variants within the gene region encoding the protein provide enhanced causal inference:
Protocol: cisMR-cML for POI Biomarker Validation The constrained maximum likelihood method for cis-MR (cisMR-cML) offers robustness to invalid instrumental variables and accounts for linkage disequilibrium among cis-SNPs [57]. Implementation involves:
Step 1: Variant Selection Identify all conditionally independent SNPs in the cis-region (typically ±500kb from transcription start site) of the candidate gene using GCTA-COJO analysis, including variants associated with either the exposure (protein level) or outcome (POI) at P < 5×10⁻⁸.
Step 2: LD Matrix Estimation Estimate the linkage disequilibrium structure among selected variants using a population-matched reference panel.
Step 3: Conditional Effect Estimation Convert marginal GWAS effects to conditional effects using the estimated LD matrix to account for correlation between variants.
Step 4: Model Fitting Apply the cisMR-cML algorithm with data perturbation to obtain causal effect estimates and standard errors robust to invalid instruments and pleiotropy.
Step 5: Bayesian Information Criterion Use BIC to consistently select the number of invalid IVs and identify valid instruments for causal inference.
This approach is particularly valuable for POI drug target discovery, as demonstrated in applications to coronary artery disease that identified potential therapeutic targets including PCSK9 [57].
Application to POI: Key Findings and Biological Insights
Inflammatory Pathways in POI Pathogenesis
PheWAS-MR analyses have revealed significant involvement of inflammatory processes in POI etiology. A recent MR study investigating 91 inflammation-related proteins identified several potential causal factors for POI [33]. The analysis employed stringent significance thresholds (P < 1×10⁻⁴ after Bonferroni correction) and validated findings through multiple sensitivity analyses.
Table 2: Inflammation-Related Proteins with Causal Effects on POI Identified via MR
| Protein | Gene | Effect Direction on POI Risk | MR P-value | Proposed Mechanism |
|---|---|---|---|---|
| CXCL10 | CXCL10 | Protective | < 1×10⁻⁴ | Chemokine signaling in ovarian tissue |
| CX3CL1 | CX3CL1 | Protective | < 1×10⁻⁴ | Fractalkine-mediated immune regulation |
| IL-18R1 | IL18R1 | Risk-increasing | < 1×10⁻⁴ | Pro-inflammatory cytokine signaling |
| MCP-1 | CCL2 | Risk-increasing | < 1×10⁻⁴ | Monocyte recruitment & activation |
| TGF-β1 | TGFB1 | Protective | < 1×10⁻⁴ | Regulation of follicular development |
The study further identified the oncostatin M signaling pathway as a potential convergent mechanism, with multiple candidate proteins (MCP-1/CCL2, TGFB1, ARTN, LIFR) implicated in this pathway [33]. Gene-drug interaction analysis prioritized CCL2 and TGFB1 as potential therapeutic targets, with genistein and melatonin identified as potential therapeutic agents for POI treatment [33].
Multi-Omics Biomarkers for POI Prediction
Integrating PheWAS-MR across multiple omics layers has revealed novel non-invasive biomarkers for POI warning. A comprehensive MR analysis incorporating metabolomic, proteomic, gut microbiome, immunophenotype, and microRNA data identified several classes of potential warning markers [34]:
Metabolomic Factors: Sphinganine-1-phosphate levels, X-23636 levels, and 4-methyl-2-oxopentanoate levels showed causal relationships with POI risk, implicating sphingolipid metabolism and branched-chain amino acid catabolism in ovarian reserve maintenance.
Circulating Plasma Proteins: Fibroblast growth factor 23 (FGF-23) and neurotrophin-3 (NT-3) levels demonstrated potential causal effects, suggesting roles in follicular development and ovarian aging.
MicroRNA Regulators: Twenty-three circulating miRNAs were identified as potential causal factors, including miR-145-5p, miR-23a-3p, and miR-221-3p, which collectively influence pathways such as glutathione metabolism and PI3 kinase signaling that are critical for ovarian function.
Immunophenotypic Markers: HVEM expression on naive CD8+ T cells emerged as a potential immune-related risk factor, highlighting the intersection between immune system function and ovarian aging.
This multi-omics PheWAS-MR approach facilitated the construction of protein-protein interaction networks that identified ESR1, ERBB2, and GART as hub genes in POI pathogenesis, providing potential targets for therapeutic intervention [34].
Implementing robust PheWAS-MR studies for POI research requires leveraging specialized analytical tools, databases, and reporting frameworks. The following table summarizes key resources that facilitate rigorous application of these methods.
Table 3: Essential Research Resources for POI-Focused PheWAS-MR Studies
| Resource Category | Specific Tools/Databases | Key Functions | Application in POI Research |
|---|---|---|---|
| Statistical Software Packages | TwoSampleMR (R), MRBase, cisMR-cML | MR analysis implementation, data harmonization, sensitivity analyses | Primary MR analysis, pleiotropy-robust estimation |
| GWAS Summary Data Platforms | EpiGraphDB, GWAS Catalog, FinnGen | Exposure and outcome data sourcing, phenotype-wide instrument selection | Access to POI GWAS statistics, multi-trait instruments |
| Reporting Guidelines | STROBE-MR checklist | Comprehensive study reporting, methodological transparency | Ensuring complete reporting of MR design and limitations |
| Colocalization Tools | PWCoCo, COLOC | Distinguishing causal associations from LD confounding | Validating protein-POI and metabolite-POI associations |
| Biological Interpretation Resources | StringDB, KEGG, miEAA | Pathway analysis, network construction, functional annotation | Interpreting multi-omics findings in POI context |
Successful application of these resources requires adherence to emerging best practices in the field, including the use of the STROBE-MR reporting guidelines to ensure comprehensive methodological transparency [59] [60]. Additionally, leveraging platforms such as the EpiGraphDB PheWAS-MR portal enables researchers to systematically explore putative causal relationships across the phenome while accounting for genetic confounding through colocalization analysis [56].
For drug target discovery applications in POI, specialized cis-MR methods such as cisMR-cML offer enhanced robustness to invalid instruments and pleiotropy [57]. These methods are particularly valuable when investigating protein biomarkers or candidate therapeutic targets encoded by specific genes, as they properly account for linkage disequilibrium among cis-SNPs and model conditional rather than marginal genetic effects.
PheWAS-MR represents a powerful framework for advancing POI research beyond single-gene investigations toward a comprehensive understanding of the complex network of causal factors contributing to disease pathogenesis. By systematically interrogating thousands of exposure-outcome relationships while leveraging genetic instruments to minimize confounding, this approach has identified novel inflammatory pathways, metabolic regulators, and potential therapeutic targets for this clinically challenging condition.
Future applications in POI research would benefit from several methodological advancements. First, increasing sample sizes in POI GWAS will enhance statistical power to detect modest causal effects. Second, integration of single-cell omics data could reveal cell-type-specific causal mechanisms in ovarian tissue. Third, application of transcriptomic and epigenomic MR methods could illuminate regulatory mechanisms underlying identified associations. Finally, developing MR methods that account for time-varying exposures could better model the progressive nature of ovarian aging.
As PheWAS-MR continues to evolve, its integration with experimental validation in model systems and triangulation with evidence from other study designs will be essential for translating statistical associations into clinically actionable insights for POI prediction, prevention, and treatment.
Navigating Analytical Pitfalls in Drug-Target MR for POI
Horizontal pleiotropy occurs when a genetic variant influences the outcome through multiple independent biological pathways, rather than solely through the exposure of interest. This phenomenon represents a fundamental violation of the Mendelian randomization (MR) "exclusion restriction" assumption, which requires that instrumental variables (IVs) affect the outcome exclusively via the exposure [61] [62]. In practical terms, horizontal pleiotropy can introduce severe biases in causal effect estimates, potentially distorting effect sizes by ranges from -131% to 201% and generating false positive causal relationships in up to 10% of MR tests [61]. The pervasiveness of horizontal pleiotropy is increasingly recognized, with studies detecting it in over 48% of significant causal relationships in MR analyses [61].
Understanding and addressing horizontal pleiotropy is particularly crucial in studies of premature ovarian insufficiency (POI) causal genes, where genetic variants often exhibit complex biological effects across multiple physiological systems. The confounded relationships between inflammatory markers, reproductive aging, and POI risk exemplify why robust pleiotropy detection methods are essential for valid causal inference [27] [63]. This protocol provides comprehensive methodologies for detecting, quantifying, and addressing horizontal pleiotropy to strengthen causal inference in MR studies of POI and related reproductive traits.
Quantifying Horizontal Pleiotropy: Statistical Frameworks and Scores
The HOrizontal Pleiotropy Score (HOPS)
The HOPS framework provides a quantitative approach to measure horizontal pleiotropy using genome-wide association study (GWAS) summary statistics. HOPS generates two distinct component scores: the pleiotropy magnitude score (Pm), which quantifies the total pleiotropic effect size of a variant across all traits, and the pleiotropy number of traits score (Pn), which measures the number of distinct pleiotropic effects a variant exhibits [64]. These scores are calculated through a statistical whitening procedure that removes correlations between traits caused by vertical pleiotropy and normalizes effect sizes across all traits. The resulting scores are scaled to represent values as they would be measured in a dataset of 100 traits, with LD-corrected versions (( {P}m^{\mathrm{LD}} ) and ( {P}n^{\mathrm{LD}} )) available to account for linkage disequilibrium [64].
HOPS can calculate both theoretical P values (based on a null scenario where variants lack pleiotropic effects) and empirical P values (corrected for polygenicity and LD) [64]. Simulation studies demonstrate that HOPS effectively distinguishes true horizontal pleiotropy from background polygenicity, with performance maintained across varying heritability assumptions and proportions of pleiotropic causal variants [64].
MR-PRESSO Global Test
The MR-PRESSO global test evaluates overall horizontal pleiotropy among all instrumental variables in a single MR test by comparing the observed distance of all variants from the regression line (residual sum of squares) against the expected distance under the null hypothesis of no horizontal pleiotropy [61]. This approach provides a global assessment of pleiotropic contamination within the entire set of instruments. When applied to complex traits and diseases, the MR-PRESSO global test has demonstrated controlled false positive rates (~5%) under the null hypothesis of no horizontal pleiotropy, with acceptable power to detect horizontal pleiotropy when the percentage of horizontal pleiotropic variants is ≥10% [61].
Table 1: Statistical Tests for Detecting Horizontal Pleiotropy
| Test Name | Underlying Principle | Application Context | Performance Characteristics |
|---|---|---|---|
| MR-PRESSO Global Test | Compares observed vs. expected residual sum of squares | Overall pleiotropy detection in multi-instrument MR | ~5% false positive rate; powerful with ≥10% pleiotropic variants [61] |
| Cochran's Q Test | Measures heterogeneity in causal estimates across instruments | Detection of unbalanced pleiotropy | Inflated false positive rates (5-25%); modified versions perform better [61] |
| MR-Egger Intercept Test | Tests for directional pleiotropy via regression intercept | Detection of average balanced pleiotropic effect | Requires InSIDE assumption; lower precision than IVW [61] [63] |
| HOPS Framework | Quantitative scoring of pleiotropic magnitude and trait count | Genome-wide pleiotropy assessment | Accounts for polygenicity; provides empirical p-values [64] |
Experimental Protocols for Pleiotropy Detection and Correction
MR-PRESSO Protocol for Outlier Detection and Correction
The MR-PRESSO framework provides a comprehensive approach to identify and correct for horizontal pleiotropic outliers in multi-instrument summary-level MR testing. The method consists of three components: a global test for horizontal pleiotropy, an outlier test for identifying specific pleiotropic variants, and a distortion test to evaluate significant differences in causal estimates before and after outlier correction [61].
Protocol Steps:
Input Preparation: Compile GWAS summary statistics for both exposure and outcome traits, ensuring consistent effect allele coding and alignment across datasets.
Instrumental Variable Selection: Identify genetic variants associated with the exposure at genome-wide significance (typically p < 5×10⁻⁸), clumping for linkage disequilibrium (LD) using standard parameters (r² < 0.001, distance > 10,000 kb) [5].
MR-PRESSO Global Test: Execute the global test to detect overall horizontal pleiotropy by comparing the observed distribution of residuals against the expected distribution under the null hypothesis of no pleiotropy.
MR-PRESSO Outlier Test: Identify specific horizontal pleiotropic outlier variants by comparing individual variant residuals against the expected distribution. Variants with significant outliers (after multiple testing correction) are flagged as potentially invalid instruments.
MR-PRESSO Distortion Test: Calculate the causal estimate before and after removing outlier variants identified in step 4. Test for significant differences between these estimates to determine if outlier removal meaningfully alters causal inference.
Sensitivity Analysis: Compare MR-PRESSO results with those from complementary methods (MR-Egger, weighted median, weighted mode) to assess robustness of causal estimates [61] [5].
Simulation studies indicate MR-PRESSO performs optimally when horizontal pleiotropy occurs in <50% of instruments, with ability to correct distortions in causal estimates and reduce false positive relationships [61].
MR-PRESSO Analytical Workflow
PCMR Framework for Correlated Horizontal Pleiotropy
The Pleiotropic Clustering framework for Mendelian Randomization (PCMR) addresses the challenging problem of correlated horizontal pleiotropy, where genetic variants influence both exposure and outcome through shared factors or biological pathways. Unlike uncorrelated horizontal pleiotropy, correlated horizontal pleiotropy presents particular difficulties for standard MR methods as the pleiotropic effects correlate with the variant-exposure associations [65].
Protocol Steps:
Model Specification: Apply the PCMR model which integrates both vertical pleiotropic (causal) effects (γ) and correlated horizontal pleiotropic effects (ηⁱ) into a unified correlated horizontal and vertical pleiotropic (HVP) effect: φⁱ = γ + ηⁱ [65].
Gaussian Mixture Modeling: Implement clustering of instrumental variables according to various HVP effects using a Gaussian mixture model: φⁱ ∼ q₁N(φ₁, σ²φ₁) + q₂N(φ₂, σ²φ₂) + ... + qₙN(φₙ, σ²φₙ) where qⱼ represents the proportion of each normal distribution [65].
Expectation-Maximization Algorithm: Estimate model parameters using the EM algorithm to classify IVs into distinct pleiotropy patterns.
Pleiotropy Test: Perform PCMR's pleiotropy test using bootstrapping to assess statistical differences between estimated effects across IV clusters, indicating significant correlated horizontal pleiotropy.
Causality Evaluation: Apply the Discernable Zero Modal Pleiotropy Assumption (DZEMPA) to identify the dominant IV category supporting a non-zero causal effect using a likelihood ratio test.
Biological Validation: Integrate functional genomic annotations (e.g., chromatin states, gene pathways) to validate clusters and exclude variants with likely correlated horizontal pleiotropic effects.
Simulation studies demonstrate PCMR effectively controls false positive rates even when correlated horizontal pleiotropic variants constitute 30-40% of instruments, outperforming conventional methods in such challenging scenarios [65].
Table 2: Comparison of Methods Addressing Horizontal Pleiotropy
| Method | Targeted Pleiotropy Type | Key Assumptions | Application in POI Research |
|---|---|---|---|
| MR-PRESSO | Uncorrelated horizontal pleiotropy | Pleiotropy occurs in <50% of instruments | Detected pleiotropy in inflammatory cytokine-POI relationships [61] [63] |
| MR-Egger | Balanced directional pleiotropy | InSIDE assumption | Used as sensitivity analysis in cytokine-POI MR studies [27] [63] |
| Weighted Median | Uncorrelated horizontal pleiotropy | >50% of weight from valid instruments | Secondary method in POI biomarker studies [5] [63] |
| PCMR | Correlated horizontal pleiotropy | Discernable ZEMPA | Suitable for POI-shared genetics with other traits [65] [28] |
| MR-TRYX | Pathway-specific pleiotropy | Outliers indicate alternative causal pathways | Potential for identifying novel POI risk factors [66] |
Advanced Frameworks for Exploiting Horizontal Pleiotropy
MR-TRYX: From Nuisance to Opportunity
The MR-TRYX framework represents a paradigm shift in addressing horizontal pleiotropy by treating pleiotropic outliers not merely as a nuisance, but as valuable indicators of alternative causal pathways affecting the outcome [66]. This approach systematically exploits horizontal pleiotropy to discover putative risk factors for disease through a structured process.
Protocol Steps:
Outlier Detection: Perform initial exposure-outcome MR analysis using standard methods (IVW, MR-Egger) and identify outlier instruments through multiple approaches (Cook's distance, Studentized residuals, heterogeneity tests) [66].
Candidate Trait Scanning: Search across comprehensive GWAS summary databases to systematically identify other traits (candidate traits) associated with the outlier variants.
Multi-Trait Pleiotropy Modeling: Develop a multi-trait model explaining heterogeneity in the exposure-outcome analysis through pathways involving candidate traits.
Outlier Adjustment: Adjust original SNP-outcome estimates for putative influences operating through candidate traits, reducing heterogeneity without complete outlier removal.
Pathway Validation: Test causal effects of identified candidate traits on the outcome using independent genetic instruments.
When applied to empirical examples, MR-TRYX has successfully identified established causal pathways and uncovered novel putative causal relationships, demonstrating how horizontal pleiotropy can be exploited for biological discovery [66].
MR-TRYX Framework for Exploiting Pleiotropy
Application to POI Research: Practical Considerations
POI-Specific Analytical Challenges
Research into premature ovarian insufficiency presents unique challenges for pleiotropy assessment due to the shared genetic architecture between reproductive aging and other physiological systems. Studies have established significant genetic correlations between age at menopause, early menopause, POI, and various health outcomes including cardiovascular disease, osteoporosis, and type 2 diabetes [28]. This shared genetics manifests as extensive horizontal pleiotropy that must be addressed for valid causal inference.
In applied POI research, multiple methods should be implemented concurrently to assess robustness. For example, in studying the relationship between inflammatory cytokines and POI, researchers have employed IVW as the primary method with MR-Egger, weighted median, weighted mode, and MR-PRESSO as sensitivity analyses [27] [63]. This multi-method approach consistently identified specific cytokines (CCL19, IL-10, IL-17A, CCL7) with potentially causal effects on POI risk while accounting for pleiotropic bias [27].
Research Reagent Solutions
Table 3: Essential Research Reagents and Resources for Pleiotropy Analysis
| Resource Category | Specific Tools/Databases | Application in Pleiotropy Analysis |
|---|---|---|
| GWAS Summary Data | FinnGen (POI cases/controls), eQTLGen Consortium, UK Biobank, GWAS Catalog | Source of exposure and outcome associations for two-sample MR [5] [63] |
| Analytical Software | MR-PRESSO R package, HOPS (GitHub), TwoSampleMR R package, PCMR implementation | Implementation of pleiotropy detection and correction methods [61] [64] [65] |
| Bioinformatics Tools | LDlink for LD reference, Cytoscape for network visualization, Sangerbox for pathway enrichment | Functional annotation of pleiotropic variants and pathway analysis [5] [44] |
| Pleiotropy Databases | GWAS ATLAS, PheWAS Catalog, GWAS Catalog | Cataloging known variant-trait associations for pleiotropy scanning [66] |
Confronting horizontal pleiotropy requires a multifaceted analytical strategy, particularly in complex traits like POI where genetic instruments frequently influence multiple biological pathways. The protocols outlined here provide a comprehensive framework for detecting, quantifying, and addressing pleiotropic bias through both correction-based and exploitation-based approaches. Implementation of these methods as standard sensitivity analyses will strengthen causal inference in MR studies of POI and enhance the validity of conclusions regarding genetic determinants and causal risk factors. As MR methodologies continue to evolve, the systematic assessment of horizontal pleiotropy remains an essential component of rigorous causal analysis in reproductive genetics and beyond.
Mendelian randomization (MR) has emerged as a powerful epidemiological tool for inferring causal relationships between exposures and outcomes by leveraging genetic variants as instrumental variables. The summary-data-based Mendelian randomization (SMR) method integrates genome-wide association study (GWAS) data with expression quantitative trait loci (eQTL) data to test for pleiotropic associations between gene expression and complex traits. However, a significant challenge in interpreting SMR results lies in distinguishing true causal relationships from spurious associations caused by linkage disequilibrium (LD). The Heterogeneity in Dependent Instruments (HEIDI) test was developed specifically to address this critical limitation.
The HEIDI test serves as a companion heterogeneity test to SMR, designed to determine whether the observed association between gene expression and a trait is due to a single shared causal variant (consistent with causality) or multiple correlated variants in linkage disequilibrium (which would invalidate causal interpretation). This distinction is particularly crucial in genomic regions with complex LD structures, where multiple correlated variants may show associations with both exposure and outcome without genuine causal relationships. For researchers investigating premature ovarian insufficiency (POI) causal genes, the HEIDI test provides an essential methodological safeguard against false positive findings arising from LD contamination.
Theoretical Foundation and Statistical Principles
Underlying Genetic Model
The HEIDI test operates under the fundamental principle that if a single causal variant influences both gene expression and the trait of interest, then the ratio of the effects (β) of any genetic variant in LD with this causal variant on the trait (β) and on gene expression (β) should be approximately constant. This relationship can be expressed as β/β ≈ k, where k represents the causal effect of gene expression on the trait.
When multiple correlated variants in a genomic region show associations with both gene expression and a trait, but no true causal relationship exists, the ratio β/β will vary significantly across variants due to different LD patterns with the distinct causal variants. The HEIDI test capitalizes on this principle by examining heterogeneity in the effect ratios across multiple SNPs in the region.
Null and Alternative Hypotheses
The HEIDI test formalizes the following statistical hypotheses:
- Null hypothesis (H₀): All SNPs in the region share a single causal variant affecting both gene expression and the trait (consistent with causality).
- Alternative hypothesis (H₁): At least two different causal variants exist for gene expression and the trait (indicating linkage rather than causality).
The test statistic is based on the heterogeneity of the ratio estimates across multiple SNPs and follows a chi-square distribution under the null hypothesis. A significant HEIDI p-value (typically < 0.01) indicates rejection of the null hypothesis, suggesting that the observed association is likely due to linkage rather than a shared causal variant.
Experimental Protocol for HEIDI Test Implementation
Prerequisite Data Requirements
Table 1: Data Requirements for HEIDI Test Analysis
| Data Type | Specifications | Source Examples | Quality Control Measures |
|---|---|---|---|
| eQTL Summary Statistics | Cis-eQTLs (±1 Mb from TSS), P < 5×10⁻⁸, MAF > 0.01 | eQTLGen, GTEx v8, BrainMeta v2, CAGE | LD pruning (r² < 0.1), F-statistic > 10 |
| GWAS Summary Statistics | Genome-wide associations for target trait | BCAC, FinnGen, IEUGWAS | Sample size > 10,000, Imputation quality > 0.8 |
| LD Reference Panel | Population-matched genotype data | 1000 Genomes, UK Biobank | Same ancestry as summary statistics |
| Annotation Files | Gene coordinates, functional annotations | ENSEMBL, RefSeq, GENCODE | Current genome build (GRCh38 recommended) |
Step-by-Step Workflow
Step 1: Data Preparation and Harmonization
- Obtain relevant eQTL summary statistics for the tissue of interest (e.g., ovarian tissue for POI research)
- Acquire GWAS summary statistics for the outcome trait (e.g., POI diagnosis)
- Ensure consistent genome build and allele coding across datasets
- Harmonize effect alleles and remove palindromic SNPs with intermediate allele frequencies
Step 2: SMR Analysis
- Perform SMR analysis using the top associated cis-eQTL as the primary instrument
- Calculate the SMR test statistic: T = (β / SE)² ~ χ² under the null hypothesis of no association
- Apply multiple testing correction (e.g., Bonferroni, FDR) for genome-wide analyses
Step 3: HEIDI Test Implementation
- Extract all cis-eQTLs within the genomic region (typically ±100 kb to ±1 Mb from gene transcription start site)
- Exclude SNPs in very strong LD (r² > 0.9) with the top eQTL to ensure independence
- Remove SNPs with weak LD (r² < 0.05) with the top eQTL that may not adequately represent the locus
- Calculate the HEIDI test statistic using the formula:
HEIDI = Σ[(β - β × k)² / (SE² + SE² × k²)]
where β and β are the effect estimates for SNP i on the outcome and exposure, respectively, SE and SE are their standard errors, and k is the ratio estimate from the top associated eQTL
Step 4: Results Interpretation
- Apply the standard significance threshold of P-HEIDI > 0.01 to support a causal interpretation
- Consider more stringent thresholds (P-HEIDI > 0.05) for increased confidence in clinical applications
- Integrate HEIDI results with other sensitivity analyses (e.g., colocalization, MR-Egger) for robust causal inference
The following workflow diagram illustrates the complete HEIDI test procedure:
Application in POI Causal Gene Research
Implementation in Multi-omics POI Studies
In a recent comprehensive MR study investigating noninvasive markers for premature ovarian insufficiency, researchers applied the HEIDI test within an integrative multi-omics framework [34]. The study integrated POI GWAS summary statistics from the FinnGen database (comprising 542 cases and 241,998 controls) with eQTL data from the eQTLGen Consortium to identify putative functional genes involved in POI pathogenesis.
The analytical approach specifically employed SMR with HEIDI testing to distinguish causal relationships from linkage effects, with significance thresholds set at FDR-adjusted P-SMR < 0.05 and P-HEIDI > 0.05 [34]. This application demonstrated the critical role of HEIDI testing in validating potential causal genes identified through MR analysis, ensuring that only robust associations proceeding to functional validation and drug target prioritization.
Integration with Complementary Methods
For POI research, the HEIDI test is most effectively deployed as part of a comprehensive causal inference pipeline:
Table 2: Complementary Methods for Causal Inference in POI Research
| Method | Purpose | Interpretation | Key Threshold |
|---|---|---|---|
| SMR with HEIDI Test | Distinguish causality from linkage | P-HEIDI > 0.01 supports shared causal variant | P-HEIDI > 0.01 (standard), > 0.05 (stringent) |
| Bayesian Colocalization | Test for shared causal variants | PP.H4 > 0.80 indicates colocalization | PP.H4 > 0.80 |
| MR-PRESSO | Detect and correct for horizontal pleiotropy | Global test P < 0.05 indicates pleiotropy | P < 0.05 |
| MR-Egger Regression | Test for directional pleiotropy | Intercept P < 0.05 suggests pleiotropy | P < 0.05 |
The relationship between these methods in a comprehensive POI causal gene discovery pipeline is illustrated below:
Research Reagent Solutions
Table 3: Essential Research Reagents and Resources for HEIDI Test Implementation
| Resource Category | Specific Examples | Application in HEIDI Test | Access Information |
|---|---|---|---|
| eQTL Summary Data | eQTLGen Consortium (31,684 samples), GTEx v8 (multiple tissues), CAGE (2,765 participants) | Provide exposure instruments for SMR analysis | Publicly available with registration |
| GWAS Summary Statistics | FinnGen (R11 release), BCAC, UK Biobank, IEUGWAS database | Outcome data for causal inference | Publicly available or through application |
| LD Reference Panels | 1000 Genomes Project, UK10K, population-specific reference panels | Calculate LD between variants for HEIDI test | Publicly available |
| Analysis Software | SMR tool, GCTA, TwoSampleMR R package, COLOC R package | Implement SMR and HEIDI test procedures | Open source or freely available |
| Bioinformatics Tools | PLINK, LDSC, METAL, FUMA | Data processing, quality control, and meta-analysis | Open source |
Interpretation Guidelines and Limitations
Critical Interpretation Framework
When applying the HEIDI test in POI research, several interpretation guidelines should be followed:
Statistical Power Considerations: The HEIDI test requires sufficient numbers of independent cis-eQTLs within the genomic region. In regions with limited eQTLs or high LD, the test may be underpowered to detect heterogeneity.
Threshold Selection: While P-HEIDI > 0.01 is the standard threshold for supporting causality, consider more stringent thresholds (P-HEIDI > 0.05) for clinical translation or drug target prioritization.
Consistency with Other Evidence: HEIDI test results should be interpreted in the context of complementary analyses, particularly Bayesian colocalization. Discordant results between HEIDI and colocalization may indicate limited power or complex genetic architectures.
Tissue Specificity: For POI research, ensure that eQTL data from relevant tissues (e.g., ovarian tissue) are used when available, as blood eQTLs may not adequately capture tissue-specific regulatory mechanisms.
Methodological Limitations
Researchers should be aware of several limitations when implementing the HEIDI test:
Power Dependency: The test's effectiveness depends on having multiple independent instrumental variables in the genomic region, which may not be available for genes with limited cis-regulatory architecture.
LD Structure Sensitivity: In regions with extremely high LD or complex haplotype structures, the HEIDI test may produce inconclusive results.
Sample Overlap Artifacts: Unaccounted sample overlap between eQTL and GWAS datasets can inflate type I error rates.
Ancestry Considerations: LD patterns differ across ancestral populations, requiring population-matched reference panels for accurate HEIDI test implementation.
The HEIDI test represents an essential methodological component in modern Mendelian randomization studies, providing critical discrimination between genuine causal relationships and linkage artifacts. For researchers investigating the genetic architecture of premature ovarian insufficiency, proper implementation and interpretation of the HEIDI test strengthens causal inference and enhances the robustness of candidate gene identification. When integrated within a comprehensive analytical framework including colocalization and sensitivity analyses, the HEIDI test contributes significantly to the validation of potential therapeutic targets and elucidation of POI pathogenesis mechanisms. As MR methodologies continue to evolve, the HEIDI test remains a cornerstone technique for ensuring the validity of causal conclusions derived from integrative genomic analyses.
In Mendelian randomization (MR) studies, which assess the causal relationship between an exposure and a disease outcome using genetic variants as instrumental variables (IVs), instrument strength is a critical determinant of validity and reliability. Weak instrument bias occurs when the genetic variants used as instruments explain only a small proportion of the variance in the exposure, potentially biasing causal effect estimates toward the confounded observational association [67]. Within the specific research context of identifying causal genes for Primary Ovarian Insufficiency (POI), a condition characterized by premature decline of ovarian function in women under 40, avoiding this bias is paramount for accurately pinpointing genuine therapeutic targets [49].
The F-statistic serves as a key diagnostic tool to detect weak instruments. A genetic variant or set of variants is traditionally considered strong enough to mitigate substantial bias if its F-statistic exceeds 10 [68] [69]. This article provides detailed application notes and protocols for ensuring instrument strength in MR studies, with a specific focus on POI research.
Theoretical Foundations: F-statistics and Weak Instrument Bias
The Role of the F-statistic in MR
In MR, the F-statistic quantifies the strength of the association between the genetic instrumental variable(s) and the exposure of interest. It is derived from the first-stage regression of the exposure on the genetic variant(s) [68]. A higher F-statistic indicates a stronger instrument, meaning the genetic variant is a more reliable proxy for the exposure.
- Calculation: For a single genetic variant, the F-statistic is approximately ( F = \frac{R^2 \times (n-2)}{1 - R^2} ), where ( R^2 ) is the proportion of variance in the exposure explained by the variant, and ( n ) is the sample size [68]. With multiple variants, a multivariate F-statistic is calculated.
- Bias Relationship: The relative bias of the two-stage least squares (2SLS) estimator is inversely proportional to the F-statistic. In one-sample MR, weak instruments bias the estimate towards the confounded observational estimate [70] [67].
Consequences of Weak Instruments
Using weak instruments (typically defined by an F-statistic < 10) can lead to several problematic outcomes [68] [70] [69]:
- Biased Causal Estimates: The MR estimate is biased toward the null in two-sample MR (with non-overlapping samples) or toward the confounded observational estimate in one-sample MR.
- Inflated Type I Error Rates: The risk of falsely identifying a causal effect increases.
- Inaccurate Confidence Intervals: Standard error estimates can be severely underestimated, leading to over-precise and misleading results.
The following diagram illustrates the causal pathways and how weak instruments lead to biased estimates, contrasting this with a valid instrumental variable scenario.
Quantitative Data and Interpretation
Interpreting F-statistic Values
The table below summarizes the interpretation of different F-statistic values in the context of MR studies.
Table 1: Interpretation of F-statistic Thresholds in Mendelian Randomization
| F-statistic Range | Interpretation | Implication for MR Analysis |
|---|---|---|
| F < 10 | Weak Instrument | Substantial bias is likely. Causal estimates are unreliable and should be interpreted with extreme caution or the instrument should be strengthened [68] [69]. |
| F ≥ 10 | Adequate Strength | A rule-of-thumb indicating that substantial weak instrument bias is unlikely. However, this is not an absolute guarantee, and higher values are always preferable [67] [71]. |
| F > 20 - 30 | Strong Instrument | Indicates a robust instrument with a low risk of weak instrument bias, leading to more reliable causal inference [71]. |
Factors Affecting the F-statistic
The F-statistic is influenced by several key factors, which are crucial to consider when designing an MR study:
- Number of Variants (k): For a fixed ( R^2 ) and sample size, increasing the number of instruments (k) decreases the F-statistic [68]. This highlights a trade-off between using more variants to increase overall explanatory power and maintaining the average strength of each variant.
- Sample Size (n): The F-statistic increases linearly with sample size. Very large sample sizes (often >10,000) are frequently required to achieve sufficient power and instrument strength in MR studies [68].
- Variance Explained (R²): The proportion of variance in the exposure explained by the genetic instruments has a direct, positive relationship with the F-statistic.
Application in POI Research: Protocols and Workflows
Protocol for Instrument Selection and F-statistic Calculation in POI MR Studies
This protocol provides a step-by-step guide for selecting genetic instruments and calculating their strength in the context of POI research, based on methodologies from recent studies [49] [34].
Step 1: Acquire Genetic Association Data
- Obtain summary-level data for the exposure (e.g., gene expression from eQTL studies) from databases such as the GTEx Portal (ovary tissue is particularly relevant for POI) or the eQTLGen Consortium [49].
- Obtain POI outcome data from sources like the FinnGen study (e.g., R11 release with 599 cases and 241,998 controls) [49].
Step 2: Select Instrumental Variables
- Identify single nucleotide polymorphisms (SNPs) significantly associated with your exposure (e.g., gene expression). A common genome-wide significance threshold is ( P < 5 \times 10^{-8} ) [71]. For smaller discovery samples, a more lenient threshold (e.g., ( P < 1 \times 10^{-5} )) is sometimes used, but this makes testing instrument strength even more critical [34].
- Clump SNPs to ensure independence based on linkage disequilibrium (e.g., ( r^2 < 0.001 ) within a 10,000 kb window) [34].
- POI-specific consideration: Prioritize instruments derived from or validated in reproductive tissues (e.g., ovary) where possible, as they may be more biologically relevant for POI [49].
Step 3: Calculate the F-statistic For a single genetic variant, the F-statistic can be calculated from summary data using the formula: [ F = \frac{R^2 \times (n - 2)}{1 - R^2} ] where ( R^2 ) is the proportion of variance in the exposure explained by the SNP, and ( n ) is the sample size of the GWAS for the exposure. The ( R^2 ) for a single SNP can be approximated using the formula: ( R^2 = 2 \times \beta^2 \times MAF \times (1 - MAF) ), where ( \beta ) is the allele effect size and ( MAF ) is the minor allele frequency [68].
For multiple variants, the approximate F-statistic for the set of instruments is: [ F = \frac{R^2 \times (n - k - 1)}{(1 - R^2) \times k} ] where ( k ) is the number of instruments and ( R^2 ) is the cumulative variance explained.
Step 4: Evaluate Instrument Strength
- Compare the calculated F-statistic to the threshold of 10.
- If ( F < 10 ), consider strategies to strengthen the instrument (see Section 4.1).
The following workflow diagram visualizes this protocol, including quality control checks.
Case Study: Identifying Causal Genes for POI
A 2024 study aimed to identify therapeutic targets for POI by integrating GWAS with eQTL data using MR and colocalization analyses [49].
- Instrument Selection: The study identified 431 genes with available index cis-eQTL signals. SNPs were selected as instruments based on their association with gene expression in whole blood (GTEx V8, n=670), ovary (GTEx V8, n=167), or the eQTLGen consortium (n=31,684) [49].
- Strength Assessment: The strength of the instrumental variables was assessed, and those with an F-statistic > 10 were selected for the main analysis to minimize weak instrument bias [49].
- Outcome: The study identified four genes (HM13, FANCE, RAB2A, and MLLT10) significantly associated with a reduced risk of POI. Subsequent colocalization analysis provided strong evidence that FANCE (involved in DNA repair) and RAB2A (involved in autophagy regulation) are promising therapeutic targets [49]. This finding was contingent on the use of strong instruments, ensuring the robustness of the causal inference.
The Scientist's Toolkit
Research Reagent Solutions
This table lists essential resources and tools for conducting instrument strength analysis in MR studies.
Table 2: Key Resources for Instrument Strength Analysis in MR Studies
| Resource / Tool | Type | Function in Analysis | Example/Reference |
|---|---|---|---|
| GTEx Portal | Database | Provides cis-eQTL data across multiple tissues, including ovary, crucial for POI studies. | [49] |
| eQTLGen Consortium | Database | A large consortium providing cis- and trans-eQTL data from peripheral blood. | [49] |
| FinnGen | Database | Source of POI GWAS summary statistics (cases and controls). | [49] [34] |
| Two-Sample MR R Package | Software | An R package for performing MR analysis, includes functions for calculating F-statistics. | [31] |
| SMR Software | Software | Tool for Summary-data-based MR analysis, integrates GWAS and eQTL data. | [49] |
| LDlink | Web Tool | A suite of tools for investigating linkage disequilibrium (LD) and performing clumping. | - |
Strategies to Mitigate Weak Instrument Bias
If instruments are weak (F < 10), consider these strategies:
- Use a Parsimonious Instrument Set: Combining genetic variants into a fewer, stronger IVs (e.g., a weighted allele score) can alleviate weak instrument problems, even with a modest decrease in power [68].
- Increase Sample Size: If possible, utilize larger GWAS summary statistics for the exposure to boost the F-statistic.
- Alternative Estimation Methods: In one-sample MR, consider methods like limited information maximum likelihood (LIML) which can be more robust to weak instruments than 2SLS [69].
- Prioritize Biological Relevance: For POI, prioritize genetic variants known to be associated with the exposure in biologically relevant tissues (e.g., ovary) rather than simply selecting all available genome-wide significant variants [49] [71].
Advanced Considerations and Limitations
- The F>10 Rule is a Guideline, Not a Law: The rule F > 10 is a useful heuristic, but it is not infallible. Bias can still be present even when F > 10, and an instrument with F just below 10 may not be severely biased [70]. The F-statistic should be interpreted as a continuous measure of strength, where higher is always better.
- Balance is Key: There is a trade-off between the number of genetic variants used (which increases total variance explained, R²) and the average strength of each variant (which affects the F-statistic). Including too many variants, especially those with small effect sizes, can reduce the average F-statistic and increase the risk of pleiotropy [71].
- Context-Specific Thresholds: In studies with very large sample sizes, a higher threshold for the F-statistic (e.g., 20-30) may be advisable to ensure minimal bias, as even small relative biases can lead to statistically significant but misleading results [71].
Vigilant assessment of instrument strength using the F-statistic is a non-negotiable step in designing and interpreting Mendelian randomization studies. This is especially critical in the search for causal genes and drug targets for complex conditions like Primary Ovarian Insufficiency, where erroneous causal inferences can misdirect valuable research resources. By adhering to the detailed protocols and considerations outlined in this article—calculating the F-statistic, aiming for values significantly greater than 10, and employing strategies to mitigate weak instrument bias—researchers can substantially enhance the validity and reliability of their findings, thereby accelerating the discovery of genuine therapeutic targets for POI.
Within the expanding application of Mendelian randomization (MR) for investigating causal genes in Premature Ovarian Insufficiency (POI), researchers are increasingly moving beyond simple protein targets. The field faces significant methodological challenges when the exposure of interest is not a single protein but a multi-protein complex or a non-protein target [16]. These complex targets are often central to biological processes, including those governing ovarian function, yet their composite nature violates standard MR assumptions that are predicated on single, distinct gene products. This application note details these specific challenges and provides structured protocols to enhance the robustness of causal inference in POI research, leveraging the principles of drug-target MR [9] [16].
Key Challenges and Strategic Considerations
The core challenge in studying complex targets with MR lies in the accurate specification of the genetic instrument, which must reliably proxy the biological exposure. The following table summarizes the primary challenges associated with multi-protein and non-protein targets.
Table 1: Key Challenges in Mendelian Randomization for Complex Targets
| Target Type | Core Challenge | Impact on MR Validity | Example from POI Context |
|---|---|---|---|
| Multi-Protein Complexes (e.g., calcium channels) | Unequal contributions of protein subunits; complex interdependencies [16]. | Instruments pooling variants across subunit genes may represent heterogeneous biological mechanisms, violating the exclusion restriction assumption. | An ion channel critical for oocyte maturation could involve multiple subunits encoded by different genes. |
| Non-Protein Targets (e.g., metabolites, lipids) | Genetic variants act through diverse and often unknown pathways [16]. | MR estimates reflect an amalgam of mechanisms, making it difficult to pinpoint a specific, actionable therapeutic intervention. | A blood metabolite identified as a potential POI biomarker [34] may be influenced by variants in many genes with different functions. |
| Targets with No Valid Instruments | Lack of strong, specific genetic proxies for the target [16]. | The MR analysis is simply not feasible, limiting the scope of investigable targets. | Approximately one-third of approved drugs lack robust genetic instruments [16]. |
The diagram below illustrates the fundamental differences in constructing valid genetic instruments for simple versus complex targets in MR studies.
Instrument Design for Simple vs. Complex Targets
A critical strategic consideration is the use of proxy exposures. When direct genetic instruments for a complex target are infeasible, downstream biomarkers can serve as proxies. For example, variant effects on a downstream metabolite can be used to infer upstream perturbations in a protein's function, as demonstrated in studies of the glycolysis pathway and vitamin D synthesis [72]. This approach requires careful consideration of the biological pathway to ensure the proxy reliably captures the target's activity.
Methodological Solutions and Advanced Analytical Frameworks
To address the challenges of pleiotropy and invalid instruments inherent in complex target analyses, advanced MR methods are essential. The following table compares several robust methods suitable for these applications.
Table 2: Advanced MR Methods for Complex Target Analysis
| Method | Core Principle | Handles Correlated SNPs? | Advantages for Complex Targets |
|---|---|---|---|
| cisMR-cML [57] | Constrained maximum likelihood to select valid IVs from a candidate set. | Yes | Robust to invalid IVs; models conditional SNP effects, crucial for correlated variants in a gene region. |
| MR-CUE [57] | Integrates multiple GWAS data sources and accounts for correlated and uncorrelated pleiotropy. | Yes | Suitable for polygenic MR setups with many SNPs across the genome. |
| Generalized IVW/Egger [57] | Extends standard MR to account for linkage disequilibrium (LD) among SNPs. | Yes | Simple extension of common methods, but assumes all IVs are valid (IVW) or requires InSIDE assumption (Egger). |
| Drug-Target MR Framework [16] | Uses variants in or near the gene encoding a drug target to proxy its perturbation. | Varies | Directly informs drug development; success rates for genetically supported targets are higher in clinical trials [9]. |
The workflow for applying a robust method like cisMR-cML is distinct from conventional MR and involves critical steps to ensure validity.
cisMR-cML Workflow for Robust Inference
Key differentiators of this workflow are:
- Step 1: Variant Selection.
cisMR-cMLincludes SNPs that are jointly associated with either the exposure or the outcome (( \mathcal{I}X \cup \mathcal{I}Y )), contrary to the standard practice of using only exposure-associated SNPs. This helps avoid introducing pleiotropy [57]. - Step 4: Modeling Conditional Effects. The method models the conditional/joint effects of SNPs, which is statistically necessary when using correlated variants, rather than relying on marginal effects from GWAS summary statistics [57].
For target prioritization from POI GWAS loci, Bayesian data integration methods like SigNet can be employed. SigNet combines within-locus evidence (e.g., gene distance, expression quantitative trait loci/eQTL colocalization) with information shared across loci via protein-protein and gene regulatory interaction networks. This can prioritize causal genes at loci where functional information is otherwise lacking [73].
Experimental Protocols
Protocol 1: cis-MR for a Multi-Protein Complex Target
Application: Assessing the causal role of a calcium channel complex in POI risk. Background: Calcium channels are often multi-subunit complexes. Pooling genetic variants from all subunit genes as instruments may conflate distinct functions of each subunit [16].
Procedure:
- Instrument Definition:
- Identify all genes encoding the primary subunits of the complex (e.g., CACNA1C, CACNB2).
- For each gene, extract cis-SNPs (e.g., within ±100 kb) from a relevant pQTL or eQTL study. Do not pool across genes at this stage.
Instrument Validation:
MR Analysis:
- Subunit-Specific Analysis: First, conduct separate cis-MR analyses for each subunit gene using a robust method like
cisMR-cML[57]. This tests the effect of perturbing each individual subunit. - Complex-Wide Sensitivity Analysis: As a secondary analysis, use a multi-variable MR (MVMR) framework. Include the genetic instruments for all major subunits as multiple, related exposures to estimate the direct effect of each, conditional on the others [38].
- Subunit-Specific Analysis: First, conduct separate cis-MR analyses for each subunit gene using a robust method like
Interpretation:
- Concordant results across subunit-specific and MVMR analyses strengthen the evidence for the complex's role.
- Discordant results suggest that the MR signal may be driven by a specific subunit's unique function, requiring careful biological interpretation.
Protocol 2: MR with a Non-Protein Proxy Exposure
Application: Investigating the causal effect of a metabolic pathway on POI using a downstream metabolite as a proxy. Background: When a direct protein target is unavailable, a downstream metabolite can serve as a proxy exposure to infer pathway perturbation, as demonstrated in studies of vitamin D synthesis [72].
Procedure:
- Exposure and Instrument Selection:
- Obtain GWAS summary statistics for the circulating metabolite (e.g., from a metabolomics GWAS) [34].
- Select genome-wide significant SNPs ((p < 5 \times 10^{-8})) associated with the metabolite as instrumental variables.
Pleiotropy Evaluation and Analysis:
- Systematically query the PhenoScanner database to identify associations of the selected IVs with traits outside the hypothesized pathway. Manually exclude SNPs with evidence of confounding pleiotropy [38].
- Apply robust MR methods (e.g., MR-Egger, MR-cML) that are less sensitive to horizontal pleiotropy [31] [57]. Use the inverse-variance weighted (IVW) method as a primary, but not sole, analysis.
Triangulation of Evidence:
- Perform colocalization analysis to assess whether the genetic association signal for the metabolite and POI share a common causal variant, reducing the likelihood of confounding by LD [34] [57].
- Interpret the MR estimate as the effect of perturbing the upstream pathway that influences the metabolite, not necessarily the direct effect of the metabolite itself [72].
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Complex Target MR
| Reagent / Resource | Function in Analysis | Key Considerations for Complex Targets |
|---|---|---|
| pQTL/eQTL Datasets [16] [57] | Source of genetic instruments for protein or gene expression exposures. | Prefer datasets from relevant tissues (e.g., ovarian tissue for POI). Be cautious of trans-QTLs that may introduce pleiotropy. |
| LD Reference Panels (e.g., 1000 Genomes) [57] | Provides correlation structure (LD matrix) among SNPs for methods like cisMR-cML. |
Must be ancestry-matched to the GWAS summary data to avoid bias. |
| PhenoScanner / GWAS Catalog [38] | Database for screening IVs for pleiotropic associations. | Critical for manually excluding variants with associations to confounding phenotypes. |
| GCTA-COJO Tool [57] | Performs conditional & joint association analysis to select independent SNPs. | Used in cisMR-cML to identify variants jointly associated with exposure or outcome. |
| GnomAD Database [2] | Catalog of human genetic variation and constraint. | Used to filter out common variants and assess gene constraint during gene prioritization. |
| FinnGen / UK Biobank [38] [34] | Source of large-scale GWAS summary statistics for outcomes (e.g., POI). | Ensure sample overlap with exposure data is minimized or accounted for in two-sample MR. |
The Critical Importance of Tissue-Specific Instrument Selection
In Mendelian randomization (MR) studies, the selection of valid genetic instruments is the most critical component for deriving reliable causal inferences. The principle of tissue-specific instrument selection extends this fundamental requirement by recognizing that genetic variants regulating molecular traits (e.g., gene expression, protein levels) often exert their effects in a tissue-specific manner. For complex conditions like Primary Ovarian Insufficiency (POI), where ovarian tissue represents the primary site of pathology, ignoring this tissue context can lead to false positive associations or obscure genuine causal relationships. This Application Note details protocols for proper tissue-specific instrument selection within MR frameworks investigating POI, enabling researchers to uncover biologically plausible therapeutic targets with greater confidence.
Background & Significance
Mendelian Randomization Fundamentals
MR uses genetic variants as instrumental variables (IVs) to probe causal relationships between exposures and outcomes, operating under three core assumptions:
- Relevance: The IV must be strongly associated with the exposure.
- Independence: The IV should not be associated with confounders.
- Exclusion: The IV should affect the outcome only through the exposure [31].
When investigating molecular exposures like gene expression or protein abundance, these genetic instruments are typically protein quantitative trait loci (pQTLs) for proteins or expression QTLs (eQTLs) for gene expression [74] [75].
The Tissue-Specificity Imperative
Genetic regulation of molecular traits is frequently tissue-dependent. A variant influencing gene expression in blood may not affect its expression in ovarian tissue. Applying instruments derived from irrelevant tissues to POI research introduces biological misclassification and violates the relevance assumption, as these variants may not actually regulate the exposure in the disease-relevant tissue context.
Multi-tissue studies demonstrate that pQTLs and eQTLs exhibit substantial tissue-specific effects [74] [75]. For example, a transcriptome-wide MR study across 48 tissues revealed that many associations are indeed tissue-specific, with thyroid-derived gene expression showing the strongest associations with thyroid disease [75]. Similarly, partitioning BMI-associated variants by tissue origin (adipose vs. brain) revealed distinct downstream effects on cardiovascular outcomes, underscoring how tissue origin drives specific pathological mechanisms [76].
For POI, where ovarian dysfunction is central, instruments derived from reproductive tissues or disease-relevant cell types are most likely to capture biologically meaningful effects.
Protocol for Tissue-Specific Instrument Selection in POI Research
Experimental Workflow
The following diagram illustrates the complete workflow for conducting a tissue-specific MR study for POI causal gene discovery:
Stepwise Procedure
Step 1: Identify Tissue-Specific Instrumental Variables
Objective: Extract genetic instruments for molecular exposures (e.g., inflammation-related proteins, metabolites) from disease-relevant tissues.
Procedure:
- Prioritize POI-Relevant Tissues: For POI research, prioritize:
- Ovarian tissue (highest relevance)
- Reproductive tissue datasets
- Hypothalamic and pituitary tissues (for hormonal regulation)
- Adipose tissue (for metabolic influences)
Access QTL Resources:
Extract Genetic Instruments:
- Apply genome-wide significance threshold (typically ( P < 5 × 10^{-8} ))
- For studies with limited power, consider a relaxed threshold (( P < 1 × 10^{-5} )) [45]
- Record effect alleles, effect sizes, and standard errors
Table 1: Tissue-Specific QTL Resources for POI Research
| Resource | Data Type | Relevant Tissues | Sample Size | Access |
|---|---|---|---|---|
| GTEx Consortium v8 | eQTLs | 48+ tissues including ovary | 80-491 per tissue | https://gtexportal.org/ |
| Olink Target Inflammation | pQTLs | Plasma (14,824 Europeans) | 14,824 | https://www.olink.com/ |
| eQTLGen | eQTLs | Whole blood | 31,684 | https://www.eqtlgen.org/ |
| FinnGen | GWAS summary statistics | POI cases/controls | 424 cases/118,796 controls | https://www.finngen.fi/ |
Step 2: Clumping and Quality Control of IVs
Objective: Ensure independence and strength of selected instruments.
Procedure:
- Clump SNPs: Remove SNPs in linkage disequilibrium (( r^2 < 0.001 ) within 10,000 kb window) [33]
- Calculate F-statistic: ( F = (β^2 / SE^2) ) for each instrument
- Harmonize alleles: Ensure consistent effect allele reporting between exposure and outcome datasets
- Remove palindromic SNPs: Avoid strand ambiguity issues
Step 3: Perform Two-Sample MR and Sensitivity Analyses
Objective: Estimate causal effects while testing for violations of MR assumptions.
Procedure:
- Primary MR Analysis:
Sensitivity Analyses:
- MR-Egger intercept test: Assess directional pleiotropy (( P < 0.05 ) suggests pleiotropy) [33]
- Cochran's Q statistic: Evaluate heterogeneity (( P < 0.05 ) indicates heterogeneity)
- Leave-one-out analysis: Test robustness to individual influential SNPs
- MR-PRESSO: Identify and correct for horizontal pleiotropy outliers [45]
Colocalization Analysis:
Step 4: Experimental Validation of Prioritized Targets
Objective: Biologically validate MR findings using experimental models.
Procedure:
- In Vitro Modeling:
- Pathway Analysis:
- Perform functional enrichment analysis of prioritized genes/proteins
- Identify overrepresented pathways (e.g., oncostatin M signaling in POI) [33]
Application Example: Inflammation-Proteins and POI
A recent MR investigation exemplified proper tissue-specific instrument selection by analyzing 91 inflammation-related proteins for causal effects on POI [33]. The study identified both protective (CXCL10, CX3CL1) and risk-increasing (IL-18R1, IL-18, MCP-1, CCL28) proteins for POI development.
Tissue-Specific Methodology
The researchers:
- Derived genetic instruments for inflammation proteins from Olink Target Inflammation panel (14,824 European participants) [33]
- Obtained POI outcome data from FinnGen Consortium (424 cases, 118,796 controls) [33]
- Applied cis-pQTLs as primary instruments to minimize pleiotropy [74]
- Conducted experimental validation in POI cell model (CTX-treated KGN cells)
Key Findings
Table 2: Causal Inflammation-Related Proteins in POI Identified via MR
| Protein | Effect on POI | MR Method | P-value | Proposed Mechanism |
|---|---|---|---|---|
| CXCL10 | Protective | IVW, Wald ratio | < 1×10⁻⁴ | Immune regulation in ovarian tissue |
| CX3CL1 | Protective | IVW, Wald ratio | < 1×10⁻⁴ | Follicle development support |
| IL-18R1 | Risk-increasing | IVW | < 1×10⁻⁴ | Pro-inflammatory signaling |
| MCP-1/CCL2 | Risk-increasing | IVW | < 1×10⁻⁴ | Monocyte recruitment in ovary |
| IL-18 | Risk-increasing | IVW | < 1×10⁻⁴ | Inflammation amplification |
| TGF-β1 | Protective (context-dependent) | Wald ratio | < 1×10⁻⁴ | Tissue remodeling regulation |
The subsequent experimental validation confirmed MCP-1/CCL2, TGFB1, ARTN, and LIFR protein expression changes in the POI model, converging on the oncostatin M signaling pathway as a potential therapeutic target [33].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Tissue-Specific POI MR Studies
| Reagent/Resource | Function | Specifications | Example Application |
|---|---|---|---|
| KGN Cell Line | Human granulosa-like tumor cells | iCell-h298, icell bioscience | In vitro POI modeling [33] |
| Cyclophosphamide (CTX) | Chemotherapeutic agent for POI induction | 1 mg/mL, 48h treatment | Creating POI cellular model [33] |
| Olink Target Inflammation Panel | Multiplex protein quantification | 96-plex immunoassays | pQTL discovery [33] |
| Anti-MCP-1 Antibody | Protein detection in validation | 1:1000 dilution (Proteintech 29547-1-AP) | Western blot confirmation [33] |
| Anti-TGF-β1 Antibody | Protein detection in validation | 1:1000 dilution (Bioss bs-0086R) | Pathway mechanism elucidation [33] |
| FinnGen R9 Data | POI GWAS summary statistics | 424 cases, 118,796 controls | MR outcome data [33] [45] |
Troubleshooting and Technical Considerations
Common Challenges
- Limited sample sizes for tissue-specific QTL discovery, particularly for ovarian tissue
- Insufficient statistical power for trans-pQTL/eQTL detection
- Horizontal pleiotropy when using multi-tissue instruments
- Cell type heterogeneity within bulk tissue samples
Mitigation Strategies
- Use Meta-Analyzed Resources: Leverage consortia data (GTEx, eQTLGen) for improved power [75]
- Prioritize cis-QTLs: These are less prone to horizontal pleiotropy and more likely to be causal [74]
- Apply Sensitivity Analyses: Rigorous pleiotropy testing (MR-Egger, MR-PRESSO) to validate assumptions [33] [45]
- Consider Cell-Type Deconvolution: Emerging single-cell QTL resources can enhance resolution
Tissue-specific instrument selection represents a methodological imperative in MR studies of POI, moving beyond convenience sampling of easily accessible tissues (e.g., blood) to biologically relevant tissues (e.g., ovarian). The integration of multi-tissue QTL atlases, rigorous sensitivity analyses, and experimental validation creates a robust framework for identifying genuine therapeutic targets. As demonstrated in the inflammation-POI context, this approach can successfully prioritize candidates like CCL2 and TGFB1 for drug development, ultimately advancing personalized therapeutic strategies for ovarian aging and insufficiency.
From Genetic Signal to Clinical Promise: Validating MR Findings
Colocalization analysis is a powerful statistical method used in genetic epidemiology to determine whether two traits share a common causal genetic variant within a specific genomic region. When applied alongside Mendelian randomization (MR), it provides compelling evidence for shared genetic mechanisms, helping to prioritize candidate causal genes for further functional validation [77] [78]. In the context of Premature Ovarian Failure (POF), also referred to as Primary Ovarian Insufficiency (POI), this integrated approach is particularly valuable for disentangling the complex etiology of the condition and identifying bona fide therapeutic targets [79] [6].
The core principle of colocalization is to test the hypothesis that the association signals for two different traits (e.g., a specific gene's expression level and a disease like POI) in a genomic region are driven by the same underlying causal single nucleotide polymorphism (SNP). This is a critical step beyond simple genetic correlation, as it differentiates mere coincidence in genomic location from a genuine shared causal mechanism [77]. For researchers and drug development professionals, a colocalization signal significantly boosts confidence in a gene's causal role, thereby de-risking the substantial investment required for subsequent functional studies and clinical development.
Theoretical Foundation and Key Concepts
Bayesian Framework for Colocalization
Modern colocalization analyses predominantly employ a Bayesian framework to evaluate the probability of different causal models given the observed genome-wide association study (GWAS) and expression quantitative trait loci (eQTL) data. A widely adopted method, implemented in the coloc R package, calculates posterior probabilities for five distinct hypotheses [6] [78]:
- PP.H0: No association with either trait.
- PP.H1: Association with trait 1 (e.g., gene expression) only.
- PP.H2: Association with trait 2 (e.g., POI) only.
- PP.H3: Association with both traits, but with different causal variants.
- PP.H4: Association with both traits, with a single shared causal variant.
A high PP.H4 (typically > 0.8 or 0.9) provides strong evidence that the two traits colocalize, meaning they are influenced by the same genetic variant [6] [78]. This framework assumes that there is at most one causal variant per trait in the tested region, though newer methods like HyPrColoc and eCAVIAR have been developed to handle scenarios with multiple causal variants [77] [78].
Distinction from Mendelian Randomization
While both are essential tools in causal inference, Mendelian Randomization (MR) and colocalization address subtly different questions. MR is primarily used to estimate the causal effect of a modifiable exposure (or risk factor) on a disease outcome. It uses genetic variants as instrumental variables to test whether higher exposure levels cause an increase in disease risk [78].
Colocalization, in contrast, investigates whether two traits share a common causal genetic variant. It does not, by itself, establish a causal relationship between the traits but confirms that their genetic signals originate from the same precise location in the genome [78]. When used together, these methods can powerfully identify exposure-mediated genetic causal pathways to a disease. For instance, MR can establish that higher BMI causes diabetes, and colocalization can then pinpoint the specific genetic variants (e.g., in the FTO gene) that influence diabetes risk specifically through BMI-mediated pathways [78].
Table 1: Key Differences Between Mendelian Randomization and Colocalization
| Feature | Mendelian Randomization (MR) | Colocalization |
|---|---|---|
| Primary Question | Does the exposure causally influence the outcome? | Do the two traits share a causal genetic variant? |
| Underlying Logic | Instrumental variable analysis | Bayesian probability |
| Key Assumption | Instruments affect outcome only via the exposure. | Signals are fine-mapped to a specific region. |
| Typical Output | Causal estimate (Odds Ratio) | Posterior Probability (e.g., PP.H4) |
| Role in Causal Pathway | Identifies the causal link between traits | Identifies the shared genetic origin |
Experimental Protocols and Workflows
Protocol 1: Standard Two-Trait Colocalization Analysis
This protocol details the steps for performing a colocalization analysis between a molecular trait (e.g., gene expression from an eQTL study) and a complex disease (e.g., POI) using the coloc R package.
Step 1: Data Preparation and Harmonization
- Obtain summary statistics for both traits for your genomic region of interest. Ensure the data includes SNP IDs, effect alleles, other alleles, effect size estimates (beta coefficients or odds ratios), and their standard errors.
- Harmonize the datasets to ensure that:
- The effect alleles are aligned across both datasets.
- Allele frequencies are from a compatible population (e.g., all of European ancestry).
- The same genome build is used.
- Define the genomic region to be tested. This is typically a locus identified from a GWAS for your disease, extended by a fixed window (e.g., ±100 kb to ±200 kb) around the lead SNP to cover all SNPs in high linkage disequilibrium (LD) [78].
Step 2: Running the Colocalization Analysis
- Load the
colocpackage in R. - Use the
coloc.abf()function, providing it with the harmonized datasets for trait 1 (e.g., eQTL) and trait 2 (e.g., POI GWAS). - Set the prior probabilities. The default priors are often used (
p1 = 1e-4, p2 = 1e-4, p12 = 1e-5), representing the prior probability of a variant being associated with trait 1, trait 2, or both, respectively [6]. - Execute the analysis.
Step 3: Interpreting the Results
- Extract the posterior probabilities (PP.H0 to PP.H4) from the results.
- A PP.H4 > 0.8 is generally considered strong evidence for colocalization, though more stringent thresholds (e.g., > 0.9) are often applied for high-confidence findings [6] [78].
- If PP.H3 is high, it suggests distinct causal variants for the two traits in the region.
- Visually inspect the results by plotting the association p-values for both traits across the region to confirm the overlap.
Protocol 2: Integrated MR-Colocalization Analysis for Causal Gene Identification
This advanced protocol integrates MR and colocalization to establish a more robust causal link between gene expression and a disease, which is directly applicable to POI research [79] [6] [78].
Step 1: Mendelian Randomization Analysis
- Select independent, genome-wide significant SNPs (typically ( P < 5 × 10^{-8} )) from the eQTL study of your candidate gene as instrumental variables.
- Extract the associations of these instrumental SNPs with the POI outcome from the GWAS summary statistics.
- Perform a two-sample MR analysis using multiple methods (e.g., Inverse Variance Weighted, MR-Egger, Weighted Median) to test for a causal effect of the gene's expression on POI risk.
Step 2: Colocalization Analysis
- On the genes that show a significant causal effect in the MR analysis, perform a colocalization test as described in Protocol 1 within the gene's cis-regulatory window.
- This step is crucial to verify that the MR signal is not driven by linkage disequilibrium (LD) with a nearby variant that is the true causal variant for POI.
Step 3: HEIDI Test for Pleiotropy
- To rule out horizontal pleiotropy (where the genetic instrument influences POI through a pathway independent of the gene's expression), perform a heterogeneity in dependent instruments (HEIDI) test.
- A ( P )-value from the HEIDI test greater than 0.05 suggests that the association is not likely due to pleiotropy and supports a single shared causal variant [6].
Diagram 1: Integrated MR-Colocalization Analysis Workflow
Protocol 3: Multi-Trait Colocalization with HyPrColoc
For more complex analyses involving many related traits (e.g., multiple molecular QTLs or correlated biomarkers), the HyPrColoc algorithm offers a computationally efficient solution [77].
Step 1: Input Preparation
- Gather GWAS summary statistics (regression coefficients and standard errors) for all
mtraits of interest for a defined genomic region. - Ensure the summary data are from the same underlying population or that the LD structure is consistent across studies.
Step 2: Executing HyPrColoc
- Use the HyPrColoc software, which is designed for the simultaneous analysis of a large number of traits.
- The algorithm will compute the posterior probability of full colocalization (PPFC)—that all
mtraits share a single causal variant. - It will also partition traits into clusters if the locus contains multiple distinct causal variants influencing different subsets of traits.
Step 3: Result Interpretation
- A high PPFC indicates that all analyzed traits in the region likely share one causal variant.
- The clustering output identifies groups of traits that share a variant, providing insights into potential shared biological pathways.
Application in Premature Ovarian Failure Research
The integrated MR-colocalization approach has been successfully applied to identify novel therapeutic targets for POI. Recent studies leveraging large-scale biobank data have demonstrated its power.
Table 2: Candidate Causal Genes for POI Identified via MR and Colocalization Analyses
| Gene Symbol | MR Evidence | Colocalization Evidence (PP.H4) | Proposed Biological Mechanism | Druggability Assessment |
|---|---|---|---|---|
| FANCE [6] | Significant (OR < 1) | Strong (PP.H4 ≥ 0.8) | DNA repair, Fanconi anemia pathway | Preclinical/Investigational |
| RAB2A [6] | Significant (OR < 1) | Strong (PP.H4 ≥ 0.8) | Autophagy regulation, vesicle trafficking | No known drugs |
| TNXB [79] | Significant (MR & SMR) | Strong (Colocalization) | Extracellular matrix organization | Preclinical/Investigational |
| BSG [79] | Significant (MR) | Strong (Colocalization) | Cell adhesion, cyclophilin ligand | Preclinical/Investigational |
A seminal study re-analyzing data from the FinnGen consortium identified 431 genes with cis-eQTL signals for testing against POI. Following MR analysis, four genes (HM13, FANCE, RAB2A, and MLLT10) were significantly associated with a reduced risk of POI. Subsequent colocalization analysis provided strong evidence specifically for FANCE and RAB2A, highlighting them as the most promising therapeutic targets [6]. FANCE is involved in DNA repair, a critical process for maintaining the finite ovarian follicle pool, while RAB2A plays a key role in autophagy, suggesting new pathways involved in ovarian aging.
Another study using plasma proteomics data identified 14 proteins with a causal relationship to POF. Colocalization analysis further refined this list, indicating that key proteins like BSG, CCL23, FAP, and TNXB share causal variants with POF traits, providing deeper insights into the disease mechanisms and potential targets for intervention [79].
The Scientist's Toolkit
Essential Research Reagent Solutions
Table 3: Key Reagents and Resources for Colocalization Analysis
| Resource Category | Specific Examples | Function and Application |
|---|---|---|
| Summary Statistics Databases | GTEx Portal (eQTLs), eQTLGen Consortium, UK Biobank, FinnGen, GWAS Catalog | Source of genetic association data for exposure and outcome traits. |
| Analysis Software & Packages | coloc R package, SMR, HyPrColoc, eCAVIAR |
Perform statistical colocalization, MR, and multi-trait analysis. |
| LD Reference Panels | 1000 Genomes Project, Haplotype Reference Consortium (HRC) | Provide population-specific linkage disequilibrium information for accurate modeling. |
| Bioinformatics Tools | PLINK, FUMA, LocusZoom | For data quality control, clumping of SNPs, and visualization of results. |
| Druggability Databases | DrugBank, DGIdb, Therapeutic Target Database (TTD), OMIM | Assess the potential of identified candidate genes as drug targets. |
Visualization of the Colocalization Hypotheses
Diagram 2: Five Hypotheses Tested in Colocalization Analysis
Colocalization analysis serves as a critical statistical tool for proving the existence of a shared causal variant between molecular traits and complex diseases like Premature Ovarian Failure. When rigorously applied within an integrative framework that includes Mendelian randomization, it moves research beyond simple association and provides a robust evidence base for prioritizing candidate causal genes. The protocols and resources outlined herein provide a roadmap for researchers and drug developers to apply this powerful method, ultimately accelerating the identification and validation of novel therapeutic targets for POI and other complex genetic disorders. As public genomic resources continue to expand, the utility and application of colocalization analysis will only grow, offering ever-deeper insights into the genetic architecture of human disease.
The high rate of failure in drug development, with only approximately 10% of clinical programmes eventually receiving approval, represents a significant challenge for the pharmaceutical industry and biomedical research [80]. This failure cost drives the need for more reliable methods to prioritize therapeutic targets with the highest probability of clinical success. Human genetic evidence has emerged as a powerful tool for this purpose, with previous work demonstrating that drug mechanisms with genetic support have a probability of success that is 2.6 times greater than those without such support [80]. This application note details the quantitative evidence, methodological protocols, and research tools for applying genetic evidence, particularly through Mendelian randomization, to improve target selection and clinical success rates, with specific relevance to research on Premature Ovarian Insufficiency (POI) causal genes.
Quantitative Evidence: The Impact of Genetic Support on Clinical Success
Analysis of 29,476 target-indication (T-I) pairs reveals a consistent and substantial advantage for drug programmes with human genetic support across multiple therapy areas and development phases [80]. The table below summarizes key quantitative findings on how genetic evidence impacts clinical success rates.
Table 1: Impact of Genetic Evidence on Drug Development Success Rates [80]
| Metric | Value with Genetic Support | Value without Genetic Support | Relative Success (RS) |
|---|---|---|---|
| Overall Probability of Success (Phase I to Launch) | Significantly Higher | Baseline | 2.6 |
| Success by Evidence Source (OMIM) | Highest | Baseline | 3.7 |
| Success by Evidence Source (GWAS) | High | Baseline | ~2.0 |
| Success by Evidence Source (Somatic - Oncology) | High | Baseline | 2.3 |
| Therapy Area - Metabolic | High | Baseline | >3.0 |
| Therapy Area - Respiratory | High | Baseline | >3.0 |
| Therapy Area - Haematology | High | Baseline | >3.0 |
| Therapy Area - Endocrine | High | Baseline | >3.0 |
| Impact of Gene Confidence (L2G Score) | Increases with higher confidence | Baseline | Positive Correlation |
The enhancement in success probability varies by therapy area, with metabolic, respiratory, haematology, and endocrine diseases showing particularly strong relative success (RS > 3.0) [80]. This effect is most pronounced in later development phases (II and III), corresponding to the critical stages where efficacy must be demonstrated. Genetic support also increases the probability of a target-indication pair transitioning from preclinical to clinical development, especially in metabolic diseases (RS = 1.38) [80].
Further analysis of stopped clinical trials using natural language processing confirms that trials halted for negative outcomes, such as lack of efficacy, show a significant depletion of genetic support (Odds Ratio = 0.61) compared to progressing trials [81]. This underscores the value of genetic evidence in mitigating the risk of late-stage failure due to lack of efficacy.
Methodological Protocols
Core Mendelian Randomization Protocol for Causal Gene Validation
Mendelian randomization (MR) is an epidemiological method that uses genetic variants as instrumental variables to test and estimate the causal effect of a modifiable exposure (e.g., a gene or protein) on a disease outcome [82]. When applied to drug target validation, it mimics a randomized controlled trial, reducing confounding and reverse causation biases prevalent in observational studies [82].
Principle: MR relies on three core instrumental variable assumptions [82]:
- Relevance: The genetic variant(s) must be robustly associated with the exposure of interest (e.g., gene expression or protein levels).
- Independence: The genetic variant(s) must not be associated with any confounders of the exposure-outcome relationship.
- Exclusion Restriction: The genetic variant(s) must affect the outcome only through the exposure, not via other pathways (no horizontal pleiotropy).
Procedure: Two-Sample MR Workflow This protocol utilizes summary-level data from two independent Genome-Wide Association Studies (GWAS) [83].
Instrument Selection:
- Data Source: Obtain summary statistics from a GWAS on the exposure (e.g., immune cell phenotypes [83] or protein quantitative trait loci [pQTL] studies).
- Criteria: Select single nucleotide polymorphisms (SNPs) associated with the exposure at a genome-wide significance threshold (typically P < 5 × 10⁻⁸) [83].
- Linkage Disequilibrium (LD): Clump SNPs to ensure independence, typically using an LD threshold of r² < 0.01 within a specified genomic window (e.g., 10,000 kb) [83].
- Strength Calculation: Calculate the F-statistic for each instrument to guard against weak instrument bias. Exclude instruments with F < 10 [83].
Outcome Data Harmonization:
- Data Source: Obtain summary statistics for the disease outcome of interest (e.g., POI) from a separate, large-scale GWAS.
- Harmonization: Align the effect alleles (EA) and other alleles (OA) for the selected SNPs between the exposure and outcome datasets. Ensure the SNP effects on both exposure and outcome are relative to the same allele.
Causal Effect Estimation:
- Perform a two-sample MR analysis using harmonized data.
- Primary Method: Use the Inverse-Variance Weighted (IVW) method as the primary analysis when multiple genetic instruments are available. This provides a reliable causal estimate under the assumption that all genetic variants are valid instruments [82].
- The IVW estimate is derived from a meta-analysis of the Wald ratio for each SNP:
β̂IVW = (Σ π̂g Γ̂g σy,g⁻²) / (Σ π̂g² σy,g⁻²)whereπ̂gis the SNP-exposure association,Γ̂gis the SNP-outcome association, andσy,gis the standard error of the SNP-outcome association [82].
Sensitivity Analyses:
- Pleiotropy Assessment: Use MR-Egger regression to test for directional pleiotropy. A significant intercept from MR-Egger suggests violation of the exclusion restriction assumption [83].
- Robustness Checks: Employ complementary methods less sensitive to pleiotropy, such as the weighted median estimator (which provides a consistent estimate if at least 50% of the weight comes from valid instruments) and mode-based estimator [83].
- Heterogeneity Test: Use Cochran's Q statistic to assess heterogeneity among the causal estimates from individual SNPs. Significant heterogeneity may indicate pleiotropy [83].
- Leave-One-Out Analysis: Iteratively remove each SNP and re-run the MR analysis to determine if the causal effect is driven by a single, potentially pleiotropic, variant.
- Directionality Test: Perform the MR-Steiger test to confirm that the causal direction is from the exposure to the outcome, and not vice versa [83].
Multiple Testing Correction:
- Account for the number of effectively independent phenotypes tested. Apply a Bonferroni correction based on this number to establish a study-wide significance threshold (e.g., P < 2.4 × 10⁻⁴ for 211 independent tests) [83]. Associations with P-values below this threshold are considered statistically significant.
The following diagram illustrates the logical relationships and workflow of this protocol.
Protocol for Integrating MR Findings with Drug Development Pipelines
To translate MR findings into actionable drug development programs, a systematic integration protocol is recommended.
Procedure:
- Trait-Indication Mapping: Map the MR-validated exposure or trait to a therapeutic indication using standardized ontologies (e.g., Medical Subject Headings [MeSH]). A high similarity threshold (e.g., ≥0.8) is recommended to define genetic support for a target-indication pair [80].
- Druggability Assessment: Evaluate the MR-validated target for druggability by:
- Querying databases like Open Targets and DrugBank for known drugs or drug classes [83].
- Assessing whether the target is a cell surface receptor, enzyme, or secreted protein amenable to therapeutic modulation.
- For the specific context of POI, investigating whether the causal gene product is a viable target for a biologic, small molecule, or other therapeutic modality.
- Evidence Integration: Triangulate MR findings with other lines of evidence:
- Observational Data: Review existing epidemiological studies for consistent associations.
- Clinical Trial Evidence: Search ClinicalTrials.gov for any existing interventions targeting the gene or pathway [83].
- Animal Models: Consider supporting evidence from genetically modified animal models, noting that human genetic evidence often shows a stronger correlation with clinical success [81].
- Safety Profiling: Investigate potential safety liabilities by:
- Examining the association of the genetic variant with other traits (phenome-wide association studies).
- Assessing gene constraint (pLI scores) in human populations; highly constrained genes may indicate that target perturbation carries safety risks [81].
- Analyzing tissue-specific expression patterns; targets with broad expression may pose higher safety risks compared to those with selective expression [81].
The Scientist's Toolkit: Research Reagent Solutions
The following table details key resources and tools essential for implementing the described Mendelian randomization and genetic evidence-based target validation protocols.
Table 2: Essential Research Resources for Genetic Evidence-Based Target Validation
| Resource Name | Type | Primary Function in Research | Relevance to POI Research |
|---|---|---|---|
| Open Targets Platform [81] | Database / Knowledge Graph | Integrates multiple data types (genetics, genomics, drugs) to rank and prioritize potential drug targets. | Identify and prioritize candidate causal genes for POI. |
| Open Targets Genetics [80] | Portal | Provides GWAS summary statistics and variant-to-gene mapping scores (L2G) for trait-associated loci. | Fine-map POI GWAS loci and assign causal genes using L2G scores. |
| TwoSampleMR R Package [83] | Software / R Package | Facilitates harmonization of exposure and outcome GWAS datasets and performs MR analyses with multiple methods. | Perform MR to test causal effects of candidate genes on POI risk. |
| GWAS Catalog | Database | Curated collection of all published GWAS, allowing discovery of genetic associations for exposures or outcomes. | Discover genetic variants associated with POI and related reproductive traits. |
| PhenoSPD [83] | Software / Tool | Decomposes phenotypic correlations to estimate the number of effectively independent tests for multiple testing correction. | Correct for multiple testing when evaluating multiple POI-related biomarkers or traits. |
| MR-Base [83] | Database / Platform | A platform that includes a database of GWAS summary data and tools for performing MR investigations. | Access pre-harmonized GWAS data for efficient MR analysis on POI. |
Application to POI Causal Genes Research
The principles and protocols outlined above can be directly applied to the investigation of Premature Ovarian Insufficiency (POI) causal genes to de-risk therapeutic development.
- From Association to Causality: Begin with known or novel genetic associations from POI GWAS. Use the MR protocol (Section 3.1) to distinguish causal genes from mere correlations. For instance, MR can test whether altered expression of a candidate gene in a specific tissue (using eQTL data) causally influences POI risk.
- Target Prioritization: Integrate MR findings for POI candidate genes with the quantitative evidence in Table 1. A gene with strong MR support for a causal role in POI has a higher prior probability of being a successful drug target. This genetic evidence should be weighted heavily when deciding which POI mechanisms to pursue preclinically.
- Clinical Trial Design: For therapeutic programs targeting a genetically validated POI gene, the increased probability of success (RS = 2.6) can inform clinical trial design and investment decisions. Furthermore, understanding that efficacy signals are most strongly differentiated in Phase II and III [80] can help in setting go/no-go milestones.
The integration of human genetics and Mendelian randomization provides a powerful, evidence-based framework for elevating POI research from gene discovery to the development of effective therapies with a substantially higher likelihood of clinical success.
Within the evolving landscape of premature ovarian insufficiency (POI) research, the integration of genetic epidemiology with traditional clinical observation has created new paradigms for causal inference. Mendelian randomization (MR) has emerged as a powerful tool for identifying potential causal factors, while retrospective cohort studies provide crucial real-world validation of these genetic findings. This methodological cross-validation is particularly valuable for POI, a condition affecting approximately 3.5% of women globally [84] [34] that remains incompletely understood despite its significant impact on fertility and overall health. This application note examines the complementary strengths of these approaches within the context of a broader thesis on Mendelian randomization for POI causal genes research, providing structured protocols and analytical frameworks for researchers investigating ovarian aging.
Comparative Analysis of Methodological Approaches
Table 1: Fundamental Characteristics of MR and Retrospective Cohort Designs in POI Research
| Characteristic | Mendelian Randomization | Retrospective Cohort Analysis |
|---|---|---|
| Core Principle | Uses genetic variants as instrumental variables to infer causality [85] | Observes existing data to identify associations between exposures and outcomes |
| Temporal Direction | Forward-time inference from genetic predisposition to outcome [85] | Backward-looking from outcome to prior exposures |
| Key Assumptions | (1) Genetic variants associate with exposure; (2) No confounding; (3) Affect outcome only through exposure [33] [42] | No unmeasured confounding; Accurate data recording; Representative sampling |
| Primary Strength | Minimizes confounding and reverse causation [85] | Reflects real-world clinical practice and population characteristics |
| POI-Specific Applications | Identifying inflammatory proteins as causal factors [33]; Discovering noninvasive warning markers [34] | Examining association between systemic sclerosis and POI risk [86]; Documenting body composition changes [87] |
| Data Sources | GWAS summary statistics [33]; Olink proteomics [33]; FinnGen database [34] | Electronic health records [86]; Clinical registries; Medical chart review |
Table 2: Exemplary Findings in POI Research Across Methodological Approaches
| Research Focus | MR Findings | Retrospective Cohort Evidence | Consistency Assessment |
|---|---|---|---|
| Inflammatory Pathways | CXCL10, CX3CL1 protective; IL-18R1, IL-18 increase risk [33] | Systemic sclerosis (autoimmune disorder) associated with 1.6x higher POI risk [86] | Supportive - both implicate immune dysfunction |
| Metabolic Factors | Identified specific metabolites including sphinganine-1-phosphate [34] | 76.9% of POI patients showed abnormal "Fat" indicators; 94.6% had elevated WHR [87] | Complementary - MR specifics mechanisms, cohort shows prevalence |
| Body Composition | Not primarily investigated in available studies | BMI significantly causally associated with age at menopause (OR=1.014) [87] | Additive - cohort establishes relationship MR could explore |
| Clinical Applications | Proposed genistein and melatonin as potential therapeutics [33] | Supports monitoring lipid metabolism and BMI in clinical management [87] | Translational - MR identifies targets, cohort informs practice |
Methodological Protocols
Protocol for Two-Sample Mendelian Randomization in POI Research
Instrumental Variable Selection
- Genetic Data Sources: Acquire GWAS summary statistics for inflammation-related proteins from studies such as the Olink Target Inflammation panel (14,824 European participants) [33]. For POI outcomes, utilize datasets from FinnGen Consortium (424 cases, 118,796 controls in R10; 542 cases, 241,998 controls in R11) [33] [34].
- SNP Selection Criteria: Apply genome-wide significance threshold (P < 5×10⁻⁸) for instrument selection. For exposures with limited instruments, consider relaxed threshold (P < 1×10⁻⁵) [34].
- Linkage Disequilibrium Management: Perform LD clustering at 10,000 kb with R² < 0.001 to ensure independence of instrumental variables [33] [42].
- Strength Validation: Calculate F-statistic for each SNP, excluding variants with F < 10 to minimize weak instrument bias [33].
MR Analysis Execution
- Primary Method: Implement inverse variance weighted (IVW) method as primary analysis approach [33] [34].
- Supplementary Methods: Apply MR-Egger, weighted median, simple mode, and weighted mode methods to assess robustness [33].
- Sensitivity Analyses:
- Conduct Cochran's Q test to quantify heterogeneity (P > 0.05 indicates no significant heterogeneity) [33].
- Perform MR-Egger intercept test to assess horizontal pleiotropy (P < 0.05 indicates potential pleiotropy) [33] [42].
- Implement MR-PRESSO global test to identify and correct for outliers [42].
- Execute "leave-one-out" analysis to evaluate if results are driven by individual SNPs [33].
Multiple Testing Correction
- Significance Thresholds: For inflammatory proteins, define significance at P < 1×10⁻⁴; for POI complications, use adjusted P < 1×10⁻³ [33].
- False Discovery Rate: Apply FDR correction (PFDR < 0.05 considered statistically significant) [42].
Protocol for Retrospective Cohort Analysis in POI Research
Cohort Definition and Eligibility
- Data Source Identification: Utilize large clinical databases such as TriNetX (US Collaborative Network), containing records for over 61 million female patients [86].
- Case Ascertainment: Define POI cases according to established diagnostic criteria, typically including amenorrhea/oligomenorrhea before age 40 with elevated FSH >25 IU/L [84] [42].
- Cohort Matching: Implement propensity score matching (PSM) to balance demographic characteristics, medications, and comorbidities between POI and control groups at a 1:1 ratio [86].
- Covariate Adjustment: Account for factors including age, BMI, smoking status, and relevant medical history that may influence ovarian function.
Statistical Analysis
- Primary Analysis: Calculate hazard ratios (HR) with 95% confidence intervals using Cox proportional hazards models to compare POI incidence between exposed and unexposed cohorts [86].
- Stratified Analyses: Perform subgroup analyses by age categories (e.g., 20-30, 30-40 years), race/ethnicity, and other relevant demographic factors [86].
- Model Diagnostics: Assess proportional hazards assumption using Schoenfeld residuals and log-log plots.
Integration of Findings and Biological Mechanisms
The convergence of evidence from MR and retrospective cohort studies provides compelling insights into POI pathogenesis, particularly regarding inflammatory pathways. MR analyses have identified specific inflammatory proteins with causal roles in POI, including protective effects of CXCL10 and CX3CL1, and risk-increasing effects of IL-18R1, IL-18, MCP-1, and CCL28 [33]. These findings align with cohort studies demonstrating increased POI risk in systemic sclerosis patients [86], supporting the involvement of immune dysregulation in ovarian aging.
Experimental validation in POI models has demonstrated significant changes in MCP-1/CCL2, TGFB1, ARTN, and LIFR, which converge in the oncostatin M signaling pathway [33]. Gene-drug interaction analyses have further identified CCL2 and TGFB1 as potential therapeutic targets, with genistein and melatonin prioritized as potential interventions [33].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for POI Mechanistic Studies
| Reagent/Cell Line | Specification | Research Application | Evidence |
|---|---|---|---|
| KGN Cell Line | Human granulosa-like tumor cell line (iCell-h298) | In vitro modeling of POI using cyclophosphamide treatment [33] | Experimental validation of MR findings |
| Anti-MCP-1 Antibody | Rabbit monoclonal (29547-1-AP, 1:1000) | Western blot detection of MCP-1 protein expression [33] | Protein validation in POI models |
| Anti-TGF-β1 Antibody | Rabbit polyclonal (bs-0086R, 1:1000) | Detection of TGF-β1 signaling pathway alterations [33] | Pathway analysis in ovarian aging |
| Anti-LIF-R Antibody | Rabbit polyclonal (22779-1-AP, 1:500) | Assessment of leukemia inhibitory factor receptor [33] | Inflammatory pathway studies |
| Cyclophosphamide | 1 mg/mL for 48h treatment (F403282) | Induction of POI model in KGN cells [33] | Experimental disease modeling |
| Olink Target Inflammation Panel | 91 inflammation-related proteins | Proteomic profiling for exposure data in MR studies [33] | Exposure data generation |
The cross-validation of MR results with retrospective cohort analyses represents a powerful approach for advancing POI research. MR provides robust causal inference regarding specific inflammatory proteins and biological pathways, while cohort studies establish real-world clinical relevance and epidemiological patterns. The convergence of evidence from these complementary methodologies strengthens the foundation for developing targeted interventions for premature ovarian insufficiency. Future research should prioritize prospective validation of identified biomarkers and experimental testing of proposed therapeutic targets in appropriate model systems.
The journey of PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) from genetic curiosity to validated therapeutic target represents a paradigm for genetically-informed drug development. This trajectory began with pioneering genetic discoveries that elucidated PCSK9's function in controlling LDL cholesterol (LDL-C) levels and its direct effects on cardiovascular health [88]. Individuals with gain-of-function (GOF) mutations in the PCSK9 gene were found to have significantly elevated LDL-C levels and dramatically increased risk of premature atherosclerotic cardiovascular disease (ASCVD), effectively constituting a third form of autosomal dominant familial hypercholesterolemia [88] [89]. Conversely, those carrying loss-of-function (LOF) variants exhibited reduced LDL-C levels and a corresponding 47-88% reduction in coronary artery disease risk [88] [89]. This "human genetic experiment" provided compelling evidence that lifelong inhibition of PCSK9 would likely reduce cardiovascular risk with a favorable safety profile, establishing the foundational rationale for drug development efforts.
The PCSK9 paradigm offers invaluable lessons for researchers investigating causal genes for complex disorders like premature ovarian insufficiency (POI), demonstrating how genetic insights can de-risk the expensive and time-consuming process of therapeutic development. The same Mendelian randomization approaches that validated PCSK9 as a target can be applied to POI research, where identifying causal genes and pathways remains challenging. This Application Note details the experimental frameworks and methodologies that enabled the PCSK9 success story, providing a roadmap for translating genetic discoveries into clinical applications across therapeutic areas.
Genetic and Molecular Foundations of PCSK9 Biology
PCSK9 Synthesis, Structure, and Regulation
PCSK9 is a serine protease primarily synthesized in the liver as a pre-pro-PCSK9 precursor protein consisting of four domains: a signal peptide, prodomain, catalytic domain, and cysteine/histidine-rich C-terminal domain [90]. Following translation, the protein undergoes autocatalytic cleavage in the endoplasmic reticulum, removing the signal peptide and enabling the prodomain to non-covalently associate with the catalytic domain [90]. This PCSK9-prodomain complex is essential for proper folding and transportation from the ER to the Golgi apparatus, where additional post-translational modifications occur [90]. The mature PCSK9 protein is then secreted into the bloodstream, where it circulates in three main forms: mature monomeric protein (LDL-bound), multimeric self-associated forms with potentially increased activity, and furin-cleaved inactive fragments [89].
The expression of the PCSK9 gene is transcriptionally regulated by sterol regulatory element-binding protein 2 (SREBP-2) and the liver-specific hepatocyte nuclear factor 1 alpha (HNF1A) [90]. This regulatory mechanism creates an interesting physiological relationship: statins, which inhibit HMG-CoA reductase, simultaneously upregulate LDL receptor (LDLR) expression through SREBP-2 activation while also increasing PCSK9 expression, thereby partially blunting their LDL-C-lowering efficacy [90]. This insight further supported the therapeutic potential of PCSK9 inhibition, particularly as an adjunct to statin therapy.
Mechanism of LDL Receptor Degradation
The primary physiological function of PCSK9 is to regulate the surface expression of LDL receptors (LDLR) on hepatocytes, the principal cells responsible for clearing LDL-C from the circulation [90]. The established mechanism involves secreted PCSK9 binding to the epidermal growth factor-like repeat A (EGF-A) domain of the LDLR on the hepatocyte surface [90] [89]. Following binding, the LDLR/PCSK9 complex undergoes clathrin-mediated endocytosis. Under normal conditions without PCSK9 binding, the LDLR would release its ligand in the acidic environment of the endosome and recycle back to the cell surface. However, when PCSK9 is bound, the acidic pH of the endosome strengthens the interaction between PCSK9's prodomain and the LDLR, preventing receptor recycling [90]. Instead of returning to the surface, the LDLR is trafficked to lysosomes for degradation [89]. A single PCSK9 molecule can facilitate the degradation of multiple LDL receptors through a proposed recycling mechanism, explaining how this relatively low-abundance protein can profoundly impact LDL receptor dynamics and plasma cholesterol homeostasis [88].
Table 1: Key Genetic Evidence Validating PCSK9 as a Drug Target
| Genetic Variant Type | Effect on PCSK9 Function | Impact on LDL-C | Cardiovascular Risk | Clinical Implications |
|---|---|---|---|---|
| Loss-of-function | Reduced activity | 15-28% reduction | 47-88% risk reduction | Protective effect; validates inhibition strategy |
| Gain-of-function | Enhanced activity | Significant elevation | Dramatically increased risk | Mimics familial hypercholesterolemia phenotype |
| Common variants | Moderate effects | Small reductions | Proportional risk reduction | Supports dose-response relationship |
Therapeutic Targeting Strategies and Clinical Translation
PCSK9 Inhibition Modalities
Multiple therapeutic approaches have been developed to inhibit PCSK9 function, each with distinct mechanisms of action:
Monoclonal Antibodies: Fully human monoclonal antibodies (e.g., evolocumab, alirocumab) represent the first class of PCSK9 inhibitors approved for clinical use [91]. These antibodies bind circulating PCSK9 in plasma, preventing its interaction with the LDLR [92]. Administered subcutaneously every 2-4 weeks, they reduce LDL-C by approximately 50-60% as monotherapy or when added to statin therapy [88] [89].
Small Interfering RNA (siRNA): Inclisiran employs GalNAc conjugation for targeted delivery to hepatocytes via the asialoglycoprotein receptor [92]. Once inside hepatocytes, it incorporates into the RNA-induced silencing complex (RISC), leading to catalytic degradation of PCSK9 messenger RNA and sustained reduction of PCSK9 protein synthesis [92]. This approach provides extended dosing intervals of approximately six months following initial loading doses [92].
Next-Generation Approaches: Emerging strategies include oral PCSK9 inhibitors, antisense oligonucleotides, and gene-editing technologies aimed at permanently disrupting PCSK9 function [93]. Recaticimab, a next-generation monoclonal antibody with an extended half-life, enables dosing intervals of 8-12 weeks while maintaining 48-59% LDL-C reduction [92].
Clinical Efficacy and Cardiovascular Outcomes
The clinical validation of PCSK9 inhibitors culminated in several landmark cardiovascular outcomes trials:
Table 2: Major Cardiovascular Outcomes Trials of PCSK9 Inhibitors
| Trial Name | Agent | Patient Population | LDL-C Reduction | CV Risk Reduction | Key Findings |
|---|---|---|---|---|---|
| FOURIER | Evolocumab | 27,564 ASCVD patients | 59% | 15-20% risk reduction | Significant reduction in MI, stroke, and coronary revascularization |
| ODYSSEY Outcomes | Alirocumab | 18,924 recent ACS patients | 57% | 15% risk reduction | Greater benefit in patients with baseline LDL-C ≥100 mg/dL |
| SPIRE-2 | Bococizumab | High-risk patients | NA | 21% risk reduction | Trial terminated early but showed significant benefit |
| ORION-9 | Inclisiran | Heterozygous FH patients | 47.9% | NA | Sustained LDL-C reduction with twice-yearly dosing |
Beyond LDL-C reduction, PCSK9 inhibitors modestly lower lipoprotein(a) [Lp(a)] by 20-30%, through mechanisms not fully understood but potentially involving LDL receptor-mediated clearance [89]. This additional effect may contribute to cardiovascular risk reduction, particularly as Lp(a) represents an independent risk factor with no currently approved specific pharmacotherapy.
Experimental Protocols for PCSK9 Research
Mendelian Randomization Framework for Target Validation
The Mendelian randomization (MR) approach that helped validate PCSK9 provides a template for investigating causal genes in POI research:
Protocol: Two-Sample Mendelian Randomization for Causal Inference
Instrumental Variable Selection:
- Identify genetic variants (SNPs) in or near the candidate gene that are significantly associated with the exposure (e.g., PCSK9 levels or activity)
- Apply genome-wide significance threshold (p < 5×10^(-8)) or gene-specific threshold (p < 1×10^(-5)) [94] [95]
- Ensure variants are independent (linkage disequilibrium R^2 < 0.001) and have F-statistics >10 to minimize weak instrument bias [5]
Data Source Harmonization:
Statistical Analysis:
- Primary analysis: Inverse-variance weighted (IVW) random-effects meta-analysis [5] [96]
- Sensitivity analyses: MR-Egger regression, weighted median, weighted mode
- Assess pleiotropy via MR-Egger intercept test (p < 0.05 indicates directional pleiotropy)
- Evaluate heterogeneity using Cochran's Q statistic
Result Interpretation:
- Scale effects to clinically meaningful units (e.g., per 50 mg/dL LDL-C reduction) [94]
- Calculate odds ratios for binary outcomes with 95% confidence intervals
- Consider biological plausibility and consistency across sensitivity analyses
Application to POI Research: This framework can be directly applied to investigate putative POI genes by using hormone levels, ovarian reserve markers, or molecular pathways as exposures, and POI diagnosis as the outcome, leveraging large-scale GWAS and biobank data.
In Vitro Assessment of PCSK9-LDLR Interactions
Protocol: Surface Plasmon Resonance (SPR) for Binding Affinity Measurements
Receptor Immobilization:
- Covalently immobilize recombinant LDLR EGF-A domain on CMS sensor chip via amine coupling
- Achieve target density of 5-10 kRU for optimal binding kinetics
- Include reference flow cell for background subtraction
Ligand Binding Analysis:
- Prepare serial dilutions of PCSK9 (0.5-500 nM) in HBS-EP+ running buffer
- Inject PCSK9 samples at 30 μL/min for 120s association phase
- Monitor dissociation for 300-600s
- Regenerate surface with 10 mM glycine-HCl (pH 2.0)
Data Processing:
- Subtract reference cell and buffer blank signals
- Fit sensorgrams to 1:1 Langmuir binding model
- Calculate kinetic parameters (ka, kd) and equilibrium dissociation constant (K_D)
Inhibition Studies:
- Pre-incubate PCSK9 with therapeutic antibodies (0.1-100 nM) for 30 minutes
- Assess binding inhibition relative to PCSK9 alone
- Calculate IC_50 values for competitive inhibitors
This protocol enables quantitative assessment of how genetic variants or therapeutic agents modulate the PCSK9-LDLR interaction, providing mechanistic insights relevant to both hypercholesterolemia and potential reproductive applications.
Visualization of PCSK9 Biology and Research Workflows
PCSK9-LDLR Regulatory Pathway
Figure 1: PCSK9 Synthesis, Secretion, and LDL Receptor Regulation Pathway. The diagram illustrates the intracellular processing of PCSK9 and its mechanism of action in promoting LDL receptor degradation, alongside therapeutic inhibition strategies.
Mendelian Randomization Workflow for Target Validation
Figure 2: Mendelian Randomization Framework for Causal Inference. The diagram outlines the core assumptions and analytical workflow for validating therapeutic targets through genetic instrumentation.
Table 3: Essential Research Reagents for PCSK9 and Mendelian Randomization Studies
| Category | Specific Reagents/Resources | Application | Key Features |
|---|---|---|---|
| Recombinant Proteins | Human PCSK9 (full-length) | Binding assays, functional studies | >95% purity, endotoxin-free |
| LDLR EGF-A domain | Interaction studies, SPR | Properly folded, biotinylated options | |
| Cell Lines | HepG2 hepatocytes | Cellular uptake studies | Endogenous LDLR expression |
| HEK293 with LDLR knockout | Specificity controls | CRISPR-engineered variants | |
| Antibodies | Anti-PCSK9 (therapeutic mAbs) | Neutralization assays | Evolocumab, alirocumab for reference |
| Anti-LDLR extracellular domain | Flow cytometry, Western blot | Non-blocking epitopes | |
| Genetic Resources | HapMap/1000 Genomes data | LD reference | Population-specific stratification |
| GWAS summary statistics | MR instrumental variables | Global Lipids Consortium, CKDGen | |
| Software Tools | TwoSampleMR R package | MR analysis | Multiple sensitivity methods |
| PLINK 2.0 | Genetic data quality control | LD calculation, scoring | |
| Biobanks | UK Biobank | Outcome data | Deep phenotyping, large N |
| FinnGen | Population-specific studies | Finnish heritage advantage |
Translational Applications for POI Research
The PCSK9 success story provides a robust framework for applying Mendelian randomization to identify and validate therapeutic targets for premature ovarian insufficiency. Key translational considerations include:
Genetic Prioritization: Apply MR to distinguish causal POI genes from merely associated variants, focusing on those with strong instrument variables and consistent effects across sensitivity analyses [5] [96].
Target Safety Profiling: Leverage lifelong genetic exposure to anticipate potential adverse effects of therapeutic modulation, as demonstrated by the favorable safety profile of PCSK9 inhibition predicted by loss-of-function variants [88] [89].
Biomarker Development: Identify circulating proteins, metabolites, or miRNAs that serve as causal mediators of POI risk using multi-omic MR approaches similar to those that validated PCSK9's role in LDL metabolism [5] [96].
Combination Therapy Potential: Explore genetic interactions between multiple targets to identify synergistic pathways, analogous to the enhanced cardiovascular risk reduction when combining LDL-C-lowering modalities [95].
The PCSK9 paradigm demonstrates that genetically-informed drug development significantly de-risks the therapeutic pipeline while providing a mechanistic understanding of disease pathophysiology. Applying these same principles to POI research offers the potential to identify novel therapeutic targets and advance much-needed interventions for this challenging condition.
The translation of high-throughput genetic discoveries into tangible clinical applications represents a significant challenge in modern biomedical research. This is particularly true for primary ovarian insufficiency (POI), a condition affecting ~3.7% of women under 40 characterized by diminished ovarian reserve and premature decline of ovarian function [33] [49]. The heterogeneous etiology of POI has hindered therapeutic development, with current treatments limited to symptom management through hormone replacement therapy and fertility interventions using donated oocytes [33].
Mendelian randomization (MR) has emerged as a powerful approach for causal inference in complex diseases, using genetic variants as instrumental variables to identify potential therapeutic targets while minimizing confounding biases [33] [49]. Recent MR studies have identified numerous potential causal genes, proteins, and metabolites for POI, creating an unprecedented opportunity for therapeutic development [33] [49] [5]. This protocol outlines a systematic framework for translating these MR-derived findings through validated preclinical models and into clinical trials, addressing a critical gap in reproductive medicine.
From Genetic Association to Therapeutic Hypothesis
Prioritizing MR-Derived Candidates
The initial step involves rigorous prioritization of MR-identified candidates based on causal strength, biological plausibility, and druggability. Recent studies have identified several high-value targets through multi-omics MR approaches:
Table 1: High-Priority Causal Targets for POI Identified via Mendelian Randomization
| Target Category | Specific Targets | Causal Direction | Proposed Mechanism | Supporting Evidence |
|---|---|---|---|---|
| Inflammation-Related Proteins | CXCL10, CX3CL1 | Protective | Anti-inflammatory signaling | MR analysis of 91 inflammatory proteins [33] |
| IL-18R1, IL-18, MCP-1, CCL28 | Risk | Pro-inflammatory signaling | MR analysis of 91 inflammatory proteins [33] | |
| DNA Repair & Autophagy Genes | FANCE, RAB2A | Protective | DNA damage repair, autophagic regulation | GWAS-integrated eQTL analysis [49] |
| Metabolites | Sphinganine-1-phosphate, 4-methyl-2-oxopentanoate | Causal | Metabolic pathway dysregulation | Metabolome-wide MR [5] [45] |
| Immunophenotypes | CD20 on IgD- CD24- B cells, Central Memory CD8+ T cells | Protective | Immune regulation | Bidirectional MR [42] |
Target Validation Workflow
The translation pathway from MR discovery to clinical application requires a structured workflow with multiple validation checkpoints:
Experimental Protocols for Preclinical Validation
In Vitro Functional Validation in POI Cell Models
Purpose: To validate the functional role of MR-identified targets in biologically relevant cell systems.
Materials and Reagents:
- Human granulosa-like tumor cell line (KGNs, iCell-h298) [33]
- Cyclophosphamide (CTX, 1 mg/mL) for POI modeling [33]
- RPMI 1640 culture medium [33]
- Primary antibodies for target proteins (e.g., MCP-1, TGF-β1, LIF-R) [33]
- Secondary antibodies (goat anti-mouse/rabbit IgG-HRP) [33]
- TRIzol reagent for RNA extraction [33]
- SeqHunt platform for RT-PCR [33]
Procedure:
- Cell Culture and POI Modeling:
- Culture KGN cells in RPMI 1640 medium at 37°C with 5% CO₂
- Treat cells with 1 mg/mL cyclophosphamide for 48 hours to establish in vitro POI model [33]
- Include untreated control cells for comparison
Gene Expression Analysis:
- Extract total RNA using TRIzol method
- Quantify RNA concentration using Nanodrop 2000
- Perform RT-PCR using SeqHunt platform or similar
- Analyze expression differences of target genes (e.g., MCP-1, TGFB1, ARTN, LIFR) between POI model and controls [33]
Protein Level Validation:
- Perform Western blot analysis as previously described [33]
- Use specific primary antibodies: anti-MCP-1 (1:1000), anti-LIF-R (1:500), anti-TGF-β1 (1:1000), anti-TNFSF14 (1:500), anti-ARTN (1:500)
- Use GAPDH (1:50,000) as loading control
- Visualize using appropriate HRP-conjugated secondary antibodies
Functional Assays:
- Assess cell viability and apoptosis in response to target modulation
- Evaluate steroid hormone production capabilities
- Measure response to gonadotropin stimulation
Validation Criteria: Significant alteration of target expression in POI model (p < 0.05) with functional impact on cell viability/apoptosis.
Pathway Mechanism Elucidation
Purpose: To identify the signaling pathways through which MR-validated targets influence ovarian function.
Experimental Approach:
- Bioinformatic Pathway Analysis:
- Perform KEGG pathway enrichment analysis using identified genes/proteins [33] [5]
- Utilize String database and Cytoscape software (version 3.10.3) to construct protein-protein interaction networks [5]
- Identify hub genes using multiple algorithms (MCC, degree, betweenness) in Cytohubba (Version: 0.1) [5]
- Experimental Validation:
- Modulate expression of identified targets (overexpression/knockdown)
- Measure downstream pathway activation (e.g., phosphorylated proteins)
- Validate specific pathway involvement using pharmacological inhibitors
Key Pathways Identified in Recent MR Studies:
- Oncostatin M signaling pathway (MCP-1/CCL2, TGFB1, ARTN, LIFR convergence) [33]
- PI3 kinase pathway [5]
- Glutathione metabolism [5]
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Research Reagents for POI Therapeutic Development
| Reagent/Category | Specific Examples | Function/Application | Source/Reference |
|---|---|---|---|
| Cell Lines | KGN human granulosa-like tumor cell line | In vitro POI modeling | iCell Bioscience [33] |
| POI Modeling Agent | Cyclophosphamide (CTX) | Inducing ovarian insufficiency in models | felixbio [33] |
| Antibodies | MCP-1, TGF-β1, LIF-R, TNFSF14, ARTN | Target protein detection | Proteintech, Bioss [33] |
| Database Resources | DGIdb, DrugBank, TTD | Druggability assessment | [33] [49] |
| Analysis Tools | String database, Cytoscape | PPI network construction | [5] |
| Pathway Resources | KEGG, Sangerbox | Pathway enrichment analysis | [5] |
Clinical Translation Framework
Druggability Assessment and Candidate Prioritization
Purpose: To evaluate the therapeutic potential of validated targets and identify repurposing opportunities.
Methodology:
- Systematic Druggability Assessment:
- Drug Repurposing Analysis:
- Identify existing compounds targeting validated pathways
- Prioritize compounds with favorable safety profiles for rapid clinical translation
Recent Findings:
- Gene-drug analysis identified CCL2 and TGFB1 as potential therapeutic targets [33]
- Genistein and melatonin were prioritized as potential drugs for POI treatment [33]
- FANCE and RAB2A identified as promising candidates for targeted therapy development [49]
Biomarker-Guided Clinical Trial Design
Purpose: To design efficient clinical trials using MR-identified biomarkers for patient stratification and treatment response monitoring.
Biomarker Categories:
- Diagnostic Biomarkers:
- Pharmacodynamic Biomarkers:
Trial Design Considerations:
- Enrich trial populations using diagnostic biomarkers
- Implement adaptive designs based on early biomarker signals
- Include biomarker response as secondary endpoints
The integration of Mendelian randomization with systematic preclinical validation provides a powerful framework for addressing the critical translational gap in POI therapeutic development. This protocol outlines a structured approach from initial genetic discovery through clinical application, leveraging recent advances in multi-omics MR to identify high-priority targets. The experimental methodologies detailed herein enable researchers to functionally validate these findings while the clinical translation framework facilitates the development of biomarker-enriched trials. As MR studies continue to expand in scale and resolution, this systematic approach promises to accelerate the development of targeted therapies for primary ovarian insufficiency, addressing a significant unmet need in women's health.
Conclusion
Mendelian Randomization has fundamentally advanced our understanding of Primary Ovarian Insufficiency by moving beyond association to establish causality for a growing list of genes involved in key ovarian functions. The integration of MR with multi-omics data provides a powerful, cost-effective framework for identifying and prioritizing high-confidence therapeutic targets, such as FANCE and RAB2A, thereby de-risking the drug development pipeline. Future efforts must focus on expanding diverse genomic resources, refining analytical methods to mitigate pleiotropy, and conducting MR within specific patient subgroups to fully realize the potential of human genetics in paving the way for novel, effective treatments for POI. The continued application of robust MR practices promises to unravel the remaining mysteries of POI etiology and deliver much-needed interventions to patients.
References
- 1. Changing Etiological Spectrum of Premature Ovarian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12248835/]
- 2. Landscape of pathogenic mutations in premature ovarian ... [https://www.nature.com/articles/s41591-022-02194-3]
- 3. Primary Ovarian Insufficiency: Causes, Symptoms & ... [https://my.clevelandclinic.org/health/diseases/17963-primary-ovarian-insufficiency]
- 4. Primary ovarian insufficiency: update on clinical and ... [https://www.frontiersin.org/journals/endocrinology/articles/10.3389/fendo.2024.1464803/full]
- 5. Noninvasive markers for warning premature ovarian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12150474/]
- 6. Identifying therapeutic targets for primary ovarian insufficiency ... [https://ovarianresearch.biomedcentral.com/articles/10.1186/s13048-024-01524-y]
- 7. Mendelian randomization integrating GWAS and eQTL ... [https://www.nature.com/articles/s41467-019-10936-0]
- 8. Mendelian Randomization: Concepts and Scope - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8725623/]
- 9. Concepts, Opportunities, Challenges, and Future Directions [https://globalheartjournal.com/articles/10.5334/gh.1438]
- 10. Morphology and Physiology of the Ovary - Endotext - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK278951/]
- 11. Follicle Growth and Development [https://www.glowm.com/section-view/heading/Follicle%20Growth%20and%20Development/item/288]
- 12. The role of LH in follicle development: from physiology to new ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11808943/]
- 13. Folliculogenesis: A Cellular Crosstalk Mechanism [https://www.mdpi.com/1467-3045/47/2/113]
- 14. GWAS Catalog [https://www.ebi.ac.uk/gwas/]
- 15. Mendelian randomization as a tool to inform drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10953771/]
- 16. Common pitfalls in drug target Mendelian randomization and ... [https://bmcmedicine.biomedcentral.com/articles/10.1186/s12916-024-03700-9]
- 17. Causal and Candidate Gene Variants in a Large Cohort ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9006976/]
- 18. MGA loss-of-function variants cause premature ovarian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11563689/]
- 19. Systematic analysis of Mendelian disease-associated gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10749499/]
- 20. Amenorrhea - StatPearls - NCBI Bookshelf - NIH [https://www.ncbi.nlm.nih.gov/books/NBK482168/]
- 21. Amenorrhea: A Systematic Approach to Diagnosis and ... [https://www.aafp.org/pubs/afp/issues/2019/0701/p39.html]
- 22. Current evaluation of amenorrhea: a committee opinion [https://www.asrm.org/practice-guidance/practice-committee-documents/current-evaluation-of-amenorrhea/]
- 23. Review of mendelian randomization studies on age at natural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10520463/]
- 24. Increased Burden of Rare Sequence Variants in GnRH ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7947783/]
- 25. Genome-wide association studies, Polygenic Risk Scores ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12013552/]
- 26. Associations of polygenic risk scores for major depression ... [https://www.nature.com/articles/s41380-025-03243-2]
- 27. Exploring the genetic association between inflammatory ... [https://www.sciencedirect.com/science/article/abs/pii/S1043466625000845]
- 28. Shared Genetics Between Age at Menopause, Early ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2021.676546/full]
- 29. The goldmine of GWAS summary statistics: a systematic ... [https://biodatamining.biomedcentral.com/articles/10.1186/s13040-024-00385-x]
- 30. Influence and role of polygenic risk score in the ... [https://jogh.org/2025/jogh-15-04071/]
- 31. A Guide to Understanding Mendelian Randomization Studies [https://pmc.ncbi.nlm.nih.gov/articles/PMC11833605/]
- 32. Mendelian randomization - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC7614635/]
- 33. Genetic Association and Drug Target Exploration Between ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12517199/]
- 34. Noninvasive markers for warning premature ovarian ... [https://ovarianresearch.biomedcentral.com/articles/10.1186/s13048-025-01696-1]
- 35. Relevance assumption - Mendelian randomization dictionary [https://mr-dictionary.mrcieu.ac.uk/term/relevance/]
- 36. Association of dietary preferences with primary ovarian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11662844/]
- 37. Association between gut microbiota and primary ovarian ... [https://www.frontiersin.org/journals/endocrinology/articles/10.3389/fendo.2023.1183219/full]
- 38. Mendelian randomization and genetic analyses reveal ... [https://www.nature.com/articles/s41598-025-10759-8]
- 39. Multi-omics Mendelian randomization integrating RNA-seq ... [https://hereditasjournal.biomedcentral.com/articles/10.1186/s41065-025-00562-w]
- 40. Integration of multi-omics quantitative trait loci evidence ... [https://www.nature.com/articles/s41598-025-12290-2]
- 41. Role of expression quantitative trait loci (eQTL) in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12357303/]
- 42. The correlation between primary ovarian insufficiency, sex ... [https://www.frontiersin.org/journals/endocrinology/articles/10.3389/fendo.2025.1456273/full]
- 43. Statistical methods for Mendelian randomization in genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9123217/]
- 44. Mendelian randomization reveals potential causal ... [https://www.nature.com/articles/s42003-024-06755-9]
- 45. A metabolome-wide Mendelian randomization study ... [https://ovarianresearch.biomedcentral.com/articles/10.1186/s13048-024-01486-1]
- 46. Introduction • TwoSampleMR [https://mrcieu.github.io/TwoSampleMR/articles/introduction.html]
- 47. Large-Scale Mendelian Randomization Study Reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10827252/]
- 48. Reciprocal causation mixture model for robust Mendelian ... [https://www.nature.com/articles/s41467-023-36490-4]
- 49. Identifying therapeutic targets for primary ovarian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11446024/]
- 50. Evidence-based guideline: Premature Ovarian Insufficiency [https://www.asrm.org/practice-guidance/practice-committee-documents/evidence-based-guideline-premature-ovarian-insufficiency--2024/]
- 51. Genetic association and drug target exploration between ... [https://www.dovepress.com/genetic-association-and-drug-target-exploration-between-inflammation-r-peer-reviewed-fulltext-article-JIR]
- 52. Review of mendelian randomization studies on age at ... [https://www.frontiersin.org/journals/endocrinology/articles/10.3389/fendo.2023.1234324/full]
- 53. Mendelian Randomization Analysis Reveals Causal ... [https://www.mdpi.com/2072-6643/15/19/4208]
- 54. Identifying therapeutic targets for primary ovarian insufficiency ... [https://ovarianresearch.biomedcentral.com/articles/10.1186/s13048-024-01524-y/tables/1]
- 55. Phenome-wide Mendelian randomisation analysis of ... [https://www.nature.com/articles/s41467-024-46927-z]
- 56. Phenome-wide Mendelian randomization mapping the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7610464/]
- 57. A robust cis-Mendelian randomization method with ... [https://www.nature.com/articles/s41467-024-50385-y]
- 58. Phenome-wide Mendelian randomization analysis reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12582838/]
- 59. Expectations for Mendelian randomization research in PLOS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12626271/]
- 60. Guidelines for Mendelian randomisation studies [https://diabetologia-journal.org/for-authors-and-reviewers/guidelines-for-mendelian-randomisation-studies/]
- 61. Detection of widespread horizontal pleiotropy in causal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6083837/]
- 62. Horizontal Pleiotropy - Mendelian randomization dictionary [https://mr-dictionary.mrcieu.ac.uk/term/horizontal-pleiotropy/]
- 63. The causal association between systemic inflammatory ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10502980/]
- 64. a quantitative score reveals pervasive horizontal pleiotropy in ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1844-7]
- 65. Leveraging pleiotropic clustering to address high ... [https://www.nature.com/articles/s41467-025-57912-5]
- 66. Exploiting horizontal pleiotropy to search for causal ... [https://www.nature.com/articles/s41467-020-14452-4]
- 67. Weak instrument bias - Mendelian randomization dictionary [https://mr-dictionary.mrcieu.ac.uk/term/weak-instrument/]
- 68. Power and instrument strength requirements for Mendelian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3147064/]
- 69. 12.3 Checking Instrument Validity [https://www.econometrics-with-r.org/12.3-civ.html]
- 70. Avoiding bias from weak instruments in Mendelian ... [https://pubmed.ncbi.nlm.nih.gov/21414999/]
- 71. A Guide for Selection of Genetic Instruments in Mendelian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7614590/]
- 72. Mendelian randomisation with proxy exposures [https://pubmed.ncbi.nlm.nih.gov/41004399/]
- 73. Prioritization of causal genes from genome-wide ... [https://pubmed.ncbi.nlm.nih.gov/39774334/]
- 74. Mendelian randomization and genetic colocalization infer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9746220/]
- 75. A transcriptome-wide Mendelian randomization study to ... [https://www.nature.com/articles/s41467-019-13921-9]
- 76. Harnessing tissue-specific genetic variation to dissect ... [https://www.sciencedirect.com/science/article/pii/S0002929721004687]
- 77. A fast and efficient colocalization algorithm for identifying ... [https://www.nature.com/articles/s41467-020-20885-8]
- 78. Integrative analysis of Mendelian and Bayesian... randomization [https://www.nature.com/articles/s41598-020-64493-4?error=cookies_not_supported&code=35b7888c-54c7-4de6-b35a-e52e6981d7fa]
- 79. Identification of Therapeutic Targets for Premature Ovarian Failure... [https://pubmed.ncbi.nlm.nih.gov/40883588/]
- 80. Refining the impact of genetic evidence on clinical success [https://www.nature.com/articles/s41586-024-07316-0]
- 81. Genetic factors associated with reasons for clinical trial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11387188/]
- 82. Mendelian randomization [https://en.wikipedia.org/wiki/Mendelian_randomization]
- 83. Mendelian randomization of immune cell phenotypes to ... [https://www.nature.com/articles/s41408-025-01277-x]
- 84. Evidence-based guideline: premature ovarian insufficiency†‡ [https://pubmed.ncbi.nlm.nih.gov/39647506/]
- 85. Mendelian Randomization Methods for Causal Inference [https://arxiv.org/html/2509.11519v1]
- 86. Association of systemic sclerosis with risk of premature ... [https://pubmed.ncbi.nlm.nih.gov/40973051/]
- 87. A retrospective cohort study and Mendelian randomization ... [https://pubmed.ncbi.nlm.nih.gov/40734494/]
- 88. PCSK9 Inhibitors: The Evolving Future - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11522611/]
- 89. PCSK9 inhibitors – mechanisms of action - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5079795/]
- 90. PCSK9 as a Target for Development of a New Generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8778893/]
- 91. PCSK9 Inhibitors: Mechanism of Action, Efficacy, and Safety [https://pubmed.ncbi.nlm.nih.gov/30207556/]
- 92. Next-generation PCSK9 inhibitors: clinical evidence and ... [https://www.e-jcpp.org/journal/view.php?number=116]
- 93. Advances in targeting LDL cholesterol: PCSK9 inhibitors ... [https://www.sciencedirect.com/science/article/pii/S2666667724000692]
- 94. Genetic variations in HMGCR and PCSK9 and kidney ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10407636/]
- 95. Causal relationship between drug target genes of LDL ... [https://lipidworld.biomedcentral.com/articles/10.1186/s12944-025-02502-z]
- 96. Metabolome-wide Mendelian randomization assessing the ... [https://pubmed.ncbi.nlm.nih.gov/39615787/]