Replication Studies for Candidate Genes: A Comprehensive Guide for Robust Genetic Association Research
This article provides a comprehensive framework for designing, executing, and interpreting replication studies for candidate genes, tailored for researchers and drug development professionals.
Replication Studies for Candidate Genes: A Comprehensive Guide for Robust Genetic Association Research
Abstract
This article provides a comprehensive framework for designing, executing, and interpreting replication studies for candidate genes, tailored for researchers and drug development professionals. It explores the critical role of replication in validating genetic associations, addressing the historical challenges and contentious findings in the field. The content covers foundational principles, rigorous methodological workflows, strategies for troubleshooting common pitfalls, and comparative analysis with genome-wide approaches. By integrating evidence from empirical studies and current best practices, this guide aims to enhance the reliability and translational potential of genetic research in biomedicine.
The Critical Role and Historical Context of Replication in Candidate Gene Research
In the field of human genetics, the identification of a disease-associated gene is merely the first step in a long validation process. For research on Premature Ovarian Insufficiency (POI) candidate genes, establishing a truly validated genetic finding requires robust replication studies that confirm initial discoveries and assess their clinical significance. This guide examines the framework for validating genetic findings in POI research, comparing key validation criteria and providing detailed experimental methodologies used in this specialized field.
The Validation Framework for Genetic Findings
A genetically validated finding is not a single discovery but a conclusion supported by consistent evidence across multiple independent studies. This process evaluates findings across several critical dimensions.
Table 1: Core Components of Genetic Test and Finding Validation
| Component | Definition | Key Question | Replication Study Focus |
|---|---|---|---|
| Analytical Validity | How accurately and reliably the test detects specific genetic variants [1] [2] | Can the test consistently detect whether a specific genetic variant is present or absent? [1] | Verify that laboratory methods can reproduce the initial variant detection with high precision. |
| Clinical Validity | How well the genetic variant is linked to the presence, absence, or risk of a specific disease [1] [2] | Is the variant conclusively associated with increased risk of POI? [3] | Confirm the statistical association between the variant and POI in an independent cohort. |
| Clinical Utility | Whether the test provides information useful for diagnosis, treatment, or prevention [1] [2] | Does knowledge of the variant lead to improved health outcomes? [3] | Assess whether the finding informs clinical management, counseling, or therapeutic decisions. |
The process of establishing these validation components follows a logical progression, building from technical accuracy to clinical application.
Validation Evidence Hierarchy
Quantitative Landscape of POI Candidate Genes: Known vs. Novel Findings
Recent large-scale genomic studies have dramatically expanded our understanding of the genetic architecture of Premature Ovarian Insufficiency. The comparison between established and newly discovered genes reveals important patterns for validation efforts.
Table 2: Validation Status of POI-Associated Genes from Recent Large-Scale Study
| Gene Category | Number of Genes | Cases Explained | Key Characteristics | Validation Evidence |
|---|---|---|---|---|
| Known POI-Causative Genes | 59 | 18.7% (193/1030) | Primarily involved in meiosis, HR repair, mitochondrial function [4] | Multiple independent reports, functional studies, inclusion in clinical panels |
| Novel POI-Associated Genes | 20 | Additional contribution (total 23.5%) | Roles in gonadogenesis, meiosis, folliculogenesis [4] | Significant burden of LoF variants in case-control analysis (P < 1 × 10^(-5)) |
| Genes with Higher Yield in PA | FSHR, etc. | 4.2% in PA vs. 0.2% in SA | More severe phenotype association [4] | Biallelic/multi-het variants more common in PA (8.3%) than SA (3.1%) |
The distribution of genetic findings across different biological mechanisms highlights the diverse pathways that can lead to POI when disrupted.
POI Gene Functional Distribution
Experimental Protocols for Validating POI Genetic Findings
Whole Exome Sequencing in POI Cohort Studies
The identification of novel POI genes typically employs a case-control design with comprehensive genetic analysis.
Protocol Overview:
- Cohort Selection: 1,030 unrelated POI patients meeting ESHRE criteria (amenorrhea before age 40 + FSH >25 IU/L on two occasions) compared to 5,000 controls [4]
- Exclusion Criteria: Chromosomal abnormalities, autoimmune diseases, iatrogenic causes [4]
- Sequencing Method: Whole exome sequencing using Illumina platforms with minimum 30x coverage
- Variant Filtering: Remove common variants (MAF >0.01 in gnomAD or population-matched controls) [4]
- Variant Classification: Follow ACMG guidelines with functional validation of uncertain significance variants [4]
Functional Validation of Candidate Variants
For variants of uncertain significance, functional studies are essential for establishing pathogenicity.
Gene-Specific Functional Assays:
- Homologous Repair Efficiency: Measure RAD51 foci formation or host cell reactivation assays for genes like HFM1, MCM8, MCM9 [4]
- Gene Expression Analysis: Quantitative RT-PCR to assess mRNA expression in ovarian tissue or appropriate cell lines
- Protein Function Assays: Western blotting, subcellular localization, and protein-protein interaction studies
Statistical Framework for Association Testing
Robust statistical methods are critical for distinguishing true associations from background noise.
Burden Testing Protocol:
- Compare burden of loss-of-function variants between cases and controls
- Apply sequence kernel association test (SKAT) for gene-based analysis
- Establish genome-wide significance threshold (P < 1 × 10^(-5)) [4]
- Calculate contribution yield as percentage of cases with pathogenic variants
The Scientist's Toolkit: Essential Reagents for POI Genetic Research
Table 3: Key Research Reagent Solutions for POI Gene Validation
| Reagent/Category | Specific Examples | Function in Validation Pipeline |
|---|---|---|
| Exome Capture Kits | Illumina Nextera, IDT xGen | Uniform coverage of coding regions for variant discovery [4] |
| NGS Platforms | Illumina NovaSeq, Ion Torrent | High-throughput sequencing for large cohort studies [5] |
| Variant Annotation | ANNOVAR, SnpEff, CADD | Functional prediction and pathogenicity assessment [4] |
| Functional Assay Kits | RAD51 foci detection, GTP/GDP exchange assays | Experimental validation of variant impact [4] |
| Gene Panel Solutions | Custom AmpliSeq panels | Targeted validation of candidate genes in replication cohorts [5] |
| CNV Detection | Microarray platforms, CNVpytor | Identification of copy number variations associated with POI [6] |
| Data Sharing | ENIGMA, gnomAD, ClinVar | Comparison with population frequencies and previous findings [4] |
Interpretation Framework for Replication Success
The validation of POI genetic findings requires careful consideration of multiple evidence types:
Confirmatory Evidence:
- Statistical Replication: Consistent association (same direction and comparable effect size) in independent cohort
- Functional Concordance: Experimental evidence supporting plausible biological mechanism
- Phenotypic Correlation: Genotype-phenotype relationships (e.g., PA vs. SA distinction) [4]
Discordant Results Analysis:
- Population Differences: Allele frequency variations across ethnic groups
- Technical Artifacts: Platform-specific biases or variant calling differences
- Methodological Heterogeneity: Differences in inclusion criteria or phenotypic characterization
Validating genetic findings in POI research requires a multi-dimensional approach that spans analytical precision, clinical association, and functional relevance. The framework presented here enables systematic comparison between established and novel POI genes, providing researchers with clear criteria for assessing replication success. As the field moves toward clinical application, these validation standards will ensure that genetic findings provide meaningful insights for diagnosis, counseling, and therapeutic development.
The replication crisis, a period of intense self-scrutiny across scientific fields, has revealed that many high-profile research findings are not replicable when independent labs repeat the experiments. In biomedical research, this has profound implications, potentially slowing drug development and misdirecting research resources. A landmark project by the Center for Open Science found that 54% of attempted preclinical cancer studies could not be replicated [7], while other internal surveys from industry reported even higher failure rates, with one finding that 89% of hematology and oncology results were irreproducible [7]. This guide examines key case studies from this crisis, focusing on candidate gene research, to compare robust and failed scientific approaches and extract lessons for improving research practices in drug development.
The Fall of Candidate Gene Studies in Psychiatry and Well-Being
Candidate gene studies were once a dominant approach for linking specific genetic variants to traits and diseases. Researchers would select genes based on their presumed biological relevance (their "candidate" status) and test for associations with particular conditions. However, this field has become one of the most prominent casualties of the replication crisis.
◉ Case Study: Genetic Foundations of Well-Being
For years, researchers proposed that specific genes were responsible for variations in human well-being. The serotonin transporter gene (SLC6A4), particularly its 5-HTTLPR promoter region, was a star candidate based on serotonin's known role in mood disorders [8]. Initial, smaller studies reported promising associations, suggesting that individuals with different versions of this gene experienced different levels of life satisfaction.
A comprehensive 2022 re-evaluation systematically reviewed this literature and tested these candidate genes against large-scale genetic databases, including the UK Biobank. The results were stark: no support was found for any of the previously proposed candidate genes or their interactions with the environment for well-being [8]. The only reliable genetic associations were identified through hypothesis-free genome-wide approaches, not the candidate gene method. The study strongly advised well-being researchers to abandon the candidate gene approach in favor of genome-wide methods [8].
◉ Case Study: Pharmacogenetics of Amphetamine Response
This failure to replicate extends beyond observational studies to experimental pharmacogenetics. A research group conducted a series of 12 candidate gene analyses on acute responses to amphetamine, studying genes including ADORA2A, SLC6A3 (dopamine transporter), and COMT [9]. When they attempted to replicate their own initial findings in a larger sample of over 200 new participants using identical methods, the result was clear: they were unable to replicate any of their previous findings [9]. This self-contained replication failure highlights the pervasiveness of the problem, even within the same research team.
Table 1: Summary of Candidate Gene Study Replication Failures
| Research Area | Prominent Candidate Genes | Replication Outcome | Key Reason for Failure |
|---|---|---|---|
| Subjective Well-Being | SLC6A4 (5-HTTLPR), BDNF, DRD4 [8] | No support found for any candidate genes in large-scale analysis [8] | Small sample sizes; small true effect sizes of individual genes [8] |
| Amphetamine Response | ADORA2A, SLC6A3, COMT, OPRM1 [9] | Zero out of 12 prior findings replicated in a larger sample [9] | Inadequate statistical power; multiple testing issues [9] |
| Depression & Schizophrenia | Historical candidate genes (e.g., 5-HTTLPR for depression) [8] | No robust evidence in large genomic studies [8] | Over-reliance on presumed biological function without genome-wide evidence [8] [10] |
A Shift in Paradigm: Lessons from Genome-Wide Association Studies (GWAS)
The repeated failure of candidate gene studies forced a major methodological shift in genetics. The field moved toward genome-wide association studies (GWAS), which scan the entire genome without pre-selecting specific genes. This approach revealed that most behavioral and psychological traits are influenced by hundreds to thousands of genetic variants, each with minuscule effects [8]. Detecting these tiny effects requires massive sample sizes, often in the hundreds of thousands, which is the opposite of the small-sample approach that plagued candidate gene research [8] [10]. This shift is a key lesson for other fields: abandoning simplistic models for data-driven, large-scale collaboration is essential for robust discovery.
◉ Case Study: A Contrast in Reproducibility - Fruit-Fly Immunology
Not all replication stories are negative. A massive project analyzing over 1,000 claims from fruit-fly immunity research published over 50 years found that at least 61% were verifiable [11]. This suggests that some biological research fields may have a stronger foundation of replicable findings. The high rate of verification in this field can be attributed to the use of model organisms with highly controlled genetics and environments, which reduces variability and increases the reliability of experimental outcomes.
Experimental Protocols in Replication Research
Conducting a rigorous replication study involves a meticulous process to ensure the results are comparable and meaningful. The following workflow outlines the standard protocol used in large-scale replication projects, such as those in cancer biology [12] [13].
Detailed Methodologies for Key Replication Experiments
Protocol Development and Preregistration: The first critical step is to write a detailed experimental plan, or a Registered Report, which is peer-reviewed and published before any data is collected [12] [14]. This prevents later manipulation of the hypotheses or analysis methods based on the results. The plan must specify:
- The exact experimental effects to be assessed.
- All protocols, including animal models, cell lines, drug doses, and treatment schedules.
- The primary outcome measures and the precise statistical analysis plan, including corrections for multiple comparisons [12].
Reagent Sourcing and Validation: A major hurdle in replication is obtaining the exact materials used in the original study. The Reproducibility Project: Cancer Biology found that none of the 53 papers they selected contained enough detail to repeat the experiments [13]. Replicators must:
- Request key reagents (cell lines, plasmids, model organisms) directly from the original authors, a process with only a 69% success rate [13].
- If unavailable, source from reputable repositories and perform validation tests (e.g., STR profiling for cell lines, sequencing for plasmids) to ensure identity and functionality.
In Vivo Replication Experiment (Example: Xenograft Model):
- Cell Line Preparation: Culture the relevant cell lines (e.g., A549 for lung adenocarcinoma, ACHN for renal carcinoma) under conditions matching the original study [12].
- Animal Model Generation: Implant cells into immunodeficient mice to grow xenograft tumors. Randomize mice into treatment and control groups to eliminate selection bias.
- Intervention: Administer the drug (e.g., cimetidine at 100 mg/kg) or vehicle control according to the original treatment schedule [12].
- Outcome Measurement: Monitor tumor volume regularly using calipers. The primary endpoint is often the relative change in tumor volume at a predetermined day (e.g., Day 11) [12].
- Data Analysis: Analyze the results strictly according to the preregistered plan, such as using an ANOVA to test for interaction effects between cell line and drug treatment, followed by planned pairwise comparisons with appropriate multiple-testing corrections [12].
The Scientist's Toolkit: Research Reagent Solutions
The replication crisis has underscored the critical importance of reliable and well-documented research materials. The following table details essential reagents and the best practices for their use.
Table 2: Key Research Reagents and Best Practices for Replicable Science
| Reagent / Material | Critical Function | Replication Challenge & Solution |
|---|---|---|
| Validated Cell Lines | Fundamental in vitro and xenograft model system. | Challenge: Contamination and misidentification. Solution: Obtain from authenticated repositories (e.g., ATCC); perform regular STR profiling. |
| Key Biological Reagents | Specific plasmids, antibodies, or chemical compounds for intervention. | Challenge: Often unpublished "secret sauce" not commercially available [13]. Solution: Journals should mandate deposition in repositories; authors must share upon request. |
| Genetically Defined Model Organisms | Controlled in vivo studies (e.g., Drosophila, mice). | Challenge: Genetic drift and differences in housing conditions. Solution: Use standardised strains from major suppliers; document housing and breeding conditions in detail. |
| Computable Phenotypes | For studies using real-world data (EHR/claims) [15]. | Challenge: Indications and endpoints often not easily extracted from structured data [15]. Solution: Develop and share precise algorithms for defining patient cohorts and outcomes. |
Visualization: The Shift from Candidate Gene to GWAS
The fundamental logic of genetic discovery has undergone a radical transformation, moving from a hypothesis-driven but often flawed approach to an open, data-driven one. The following diagram contrasts these two paradigms.
The high-profile failures in candidate gene research offer a clear lesson: scientific rigor must trump compelling narratives. The path forward requires a fundamental change in practice and incentives. Key solutions include:
- Embracing Large-Scale Collaboration: As genetics has shown, many complex questions can only be answered by pooling data and resources across institutions to achieve sufficient statistical power [8] [10].
- Mandating Transparency: Journals and funders must require full protocol sharing, data archiving, and detailed "failure analyses" that document the tricks and hurdles essential for making an experiment work [7] [13].
- Reforming Incentives: The scientific ecosystem must reward robust, replicated findings over novel but potentially flimsy breakthroughs. This involves supporting replication studies financially and publishing their results regardless of outcome [16] [14].
The replication crisis is not a sign of science's failure, but a painful but necessary phase of self-correction. By learning from the past and adopting more rigorous, transparent, and collaborative methods, researchers and drug developers can build a more reliable foundation for future discoveries.
Replication is a cornerstone of the scientific method, serving as a critical check on the validity and reliability of research findings. In fields like candidate gene research, where findings often form the foundation for further drug development and clinical studies, the inability to replicate results can lead to a waste of resources and misdirected scientific effort. This guide examines the indispensable role of replication in combating false positives and publication bias, providing researchers with a framework for evaluating and implementing rigorous replication practices.
The Replication Crisis and Its Core Drivers
Science is facing a “replication crisis” in which many high-profile experimental findings cannot be replicated in subsequent studies [17]. Large-scale analyses have revealed that many published papers in fields ranging from cancer biology to psychology cannot be replicated, raising concerns that many published conclusions may be false [17]. This crisis is primarily driven by two interconnected factors: publication bias and the misuse of statistical significance.
Publication bias arises when the probability that a study is published is not independent of its results [17]. A substantial majority of published scientific results are positive, with more than 80% of papers across disciplines reporting positive findings, and this number exceeds 90% in fields such as psychology and ecology [17]. This bias is starkly illustrated by research on antidepressant studies, which found that 37 of 38 positive studies were published, but only 3 of 24 negative studies were published as negative results [17].
This preference for positive findings creates a "file drawer problem," where statistically non-significant results are filed away and never published [17] [18]. Consequently, the published literature presents a distorted view of the evidence, systematically overrepresenting positive findings. When a scientific community relies on this biased body of literature, false claims can frequently become canonized as fact [17].
A Framework for Interpreting Replication Results
Interpreting the outcomes of replication studies requires moving beyond a simple "success/failure" dichotomy. Statistically sophisticated researchers often struggle with this interpretation [19]. The table below outlines key statistical outcomes and their interpretations when an original study and a replication study are compared.
Table 1: Interpretation of Replication Study Outcomes
| Original Study Result | Replication Result | Probability when H₀ is True | Probability when H₁ is True (50% power) | Interpretation & Implications |
|---|---|---|---|---|
| Significant | Significant | 0.25% (alpha²) | 25% ((1-beta)²) | Strong evidence against H₀; unlikely to occur by chance if H₀ is true. |
| Significant | Non-Significant | 4.25% (alpha*(1-alpha)) | 25% (beta*(1-beta)) | Inconsistent results; difficult to interpret without meta-analysis. |
| Non-Significant | Significant | 4.25% ((1-alpha)*alpha) | 25% ((1-beta)*beta) | Inconsistent results; difficult to interpret without meta-analysis. |
| Non-Significant | Non-Significant | 90.25% ((1-alpha)²) | 25% (beta²) | Suggests H₀ may be true, but does not prove it (absence of evidence). |
A single replication failure does not necessarily invalidate an original finding. Inconsistent results (one significant, one non-significant) are surprisingly common, especially when statistical power is low [19]. With a typical power of 50%, this outcome occurs 50% of the time whether the null hypothesis is true or false, making it largely uninformative [19]. A more powerful approach is effect size meta-analysis, which combines results from original and replication studies to provide a more accurate overall estimate [19].
Types and Methodologies of Replication Studies
Replication is not a monolithic concept. Different types of replication studies serve distinct purposes in validating scientific claims [20].
- Direct Replication: A new study follows the original study's methods as closely as possible. The goal is to determine whether the finding can be reproduced under similar conditions, though samples, specific conditions, and research teams are necessarily different [20].
- Conceptual Replication: A study employs different methodologies or measures to test the same underlying hypothesis or theoretical construct as the original study. This approach tests the robustness and generalizability of a finding beyond the specific original operationalization [20].
The distinction between "direct" and "conceptual" is increasingly seen as a continuum rather than a strict dichotomy. Both are valuable, and the choice depends on the specific research question being investigated [20].
A Protocol for Conducting Replication Research
Conducting a rigorous replication study requires careful planning and execution. The following workflow, based on guidelines from the Open Science Framework, outlines the key stages [20].
Diagram 1: Replication Study Workflow
Key steps include [20]:
- Identifying a study that is feasible to replicate given available time, expertise, and resources.
- Obtaining the original materials and protocols used in the initial study.
- Developing a detailed plan specifying the type of replication and research design.
- Implementing the study according to pre-specified best practices.
- Conducting the study, analyzing the data, and sharing the results regardless of the outcome.
When conducting replications, researchers should avoid critiquing the original study's design as a basis for the replication findings and should always communicate with the original authors throughout the process [20].
Replication in Action: A Case Study from Genetics
The critical importance of functional validation through replication is powerfully illustrated in genome-wide association studies (GWAS). GWA studies identify statistical associations between genetic markers and traits, but these associations are prone to false positives [21]. Therefore, functional validation is necessary to make strong claims about gene function [21].
A landmark study in Medicago truncatula demonstrated this principle. Researchers began with a GWAS that identified an initial list of 100 candidate single nucleotide polymorphisms (SNPs) most strongly associated with variation in nodulation [21]. This list was filtered based on statistical support, linkage disequilibrium, location near annotated genes, and expression in relevant tissues [21]. Ten candidate genes were selected for functional validation using three independent reverse genetics mutagenesis tools [21].
Table 2: Key Research Reagent Solutions for Genetic Replication
| Research Reagent / Tool | Function in Validation | Application in the Case Study |
|---|---|---|
| Tnt1 Retrotransposon Mutants | Disrupts gene function by inserting a mobile genetic element into the gene sequence. | Used to create homozygous mutant lines (e.g., pho2-likeTnt, pen3-likeTnt). |
| Hairpin RNA-Interference (RNAi) | Knocks down gene expression by triggering the degradation of specific mRNA sequences. | Generated stable whole-plant knockdown lines (e.g., pno1-likeHP). |
| CRISPR/Cas9 Nuclease | Provides precise, targeted gene editing to create specific knock-out mutations. | Used to generate eight independent mutant lines for validation. |
| Phenotypic Assays | Measures the observable characteristics (phenotypes) resulting from genetic manipulation. | Well-replicated experiments to count nodule production in mutants vs. wild types. |
The use of multiple, independent mutagenesis platforms (Tnt1, RNAi, CRISPR/Cas9) constituted a form of internal replication, where the same research team tests the hypothesis using different methods [21] [20]. The results were striking: of the ten candidate genes identified by the initial GWAS, only three (PHO2-like, PNO1-like, and PEN3-like) showed statistically significant effects on nodule production in the validation experiments, and each of these was confirmed in two independent mutants [21]. This demonstrates how rigorous replication separates true signals from statistical noise.
The Economic Value and Strategic Funding of Replication
Given limited scientific resources, a key question is how much should be allocated to replication versus new research. A data-driven framework suggests that a well-calibrated replication program could productively spend about 1.4% of the National Institutes of Health (NIH) annual budget before hitting negative returns relative to funding new science [22]. This is significantly less than some political suggestions of 20%, but still represents a substantial investment that would far exceed current levels [22].
The value of a replication study is highest when it is targeted at recent, influential studies that are likely to receive substantial downstream attention. The return on investment is driven by four key factors [22]:
- The probability the finding would be overturned by a replication.
- The amount of downstream attention the study would receive without a replication.
- The proportion of that attention preempted by a failed replication.
- The cost of replication relative to a new study.
A conservative analysis suggests that about 11% of published studies may be genuinely unreliable [22]. By shutting down these false leads early, replications prevent follow-on resources from being spent on fruitless research, thereby accelerating genuine discovery [22].
Building a More Robust Scientific Future
Combating false positives and publication bias requires a systemic shift in scientific incentives and practices. Promising reforms include [20]:
- Full transparency of materials, methods, and data.
- Study pre-registration, where researchers publicly declare their hypotheses and analysis plan before data collection begins.
- Result-blind peer review, where journals accept studies based on the importance of the research question and rigor of the methodology, not the nature of the results.
- Statistical reform, including redefining the significance of the p-value and encouraging the use of Bayesian methods, which can reduce the occurrence of publication bias [23].
Replication is a form of scientific checks and balances. It is no more or less important than other parts of the scientific method, but it is absolutely necessary to perpetuate the self-correcting cycle of scientific discovery [20]. For researchers in candidate gene research and drug development, embracing replication is not an admission of failure but a commitment to building a more reliable, efficient, and credible foundation for future innovation.
The field of genetic epidemiology has undergone a profound transformation over the past two decades, moving from hypothesis-driven candidate gene studies to comprehensive, hypothesis-generating genome-wide association studies (GWAS). This paradigm evolution represents more than just a methodological shift—it reflects a fundamental change in how researchers approach complex genetic architectures. Candidate gene studies, which focus on one or a small number of biologically presumed genes, once dominated the literature but have increasingly been supplanted by GWAS, which agnostically test hundreds of thousands to millions of genetic variants across the genome [24].
This transition was driven by growing recognition that previous biological knowledge was often insufficient to correctly specify candidate gene hypotheses, leading to high rates of false positives and low replication rates [25] [24]. Meanwhile, technological advances in high-throughput genotyping and computational methods have made GWAS increasingly accessible and powerful. The implications of this shift extend throughout biomedical research, affecting how studies are designed, how findings are validated, and how genetic discoveries are translated into clinical applications.
Fundamental Methodological Differences
Core Principles and Approaches
The fundamental distinction between these approaches lies in their starting points and underlying philosophies. Candidate gene studies begin with prior biological knowledge about gene function and presumed relevance to a phenotype, typically testing a limited number of single nucleotide polymorphisms (SNPs) in genes selected based on their understood biological roles [26] [24]. This targeted approach offers dense coverage of specific genomic regions but is inherently limited by current biological understanding.
In contrast, GWAS takes an agnostic, data-driven approach that scans the entire genome without pre-specified hypotheses about particular genes. By examining hundreds of thousands to millions of genetic markers simultaneously, GWAS can discover entirely novel genetic associations independent of prior biological knowledge [26] [25]. This comprehensive coverage comes with significant multiple testing burdens, requiring stringent statistical thresholds (typically P < 5 × 10⁻⁸) to account for the vast number of comparisons being made [26] [27].
Statistical Power and Multiple Testing Challenges
The statistical properties of these approaches differ dramatically, particularly regarding power and multiple testing corrections. The table below summarizes key comparative aspects:
Table 1: Fundamental Methodological Differences Between Approaches
| Aspect | Candidate Gene Studies | Genome-Wide Association Studies (GWAS) |
|---|---|---|
| Hypothesis Framework | Hypothesis-driven based on prior biological knowledge | Hypothesis-generating, agnostic scan |
| Genomic Coverage | Limited to preselected genes | Comprehensive genome-wide coverage |
| Number of Variants Tested | Typically 1-100 variants | 100,000 to millions of variants |
| Statistical Threshold | Standard significance (P < 0.05) | Genome-wide significance (P < 5 × 10⁻⁸) |
| Multiple Testing Burden | Minimal | Extreme, requiring stringent correction |
| Discovery Potential | Limited to known biology | Can reveal novel biological pathways |
| Primary Strength | Deep investigation of specific genes | Unbiased discovery capacity |
Statistical power represents a crucial differentiator between these approaches. Candidate gene studies tend to have greater inherent statistical power for detecting effects in their targeted regions because they test far fewer variants and thus require less stringent multiple testing corrections [26]. However, this apparent advantage is offset by a critical limitation: if the true causal genes lie outside the preselected candidates, these studies have zero power to detect them.
GWAS addresses this limitation through comprehensive genomic coverage but pays a substantial penalty in multiple testing. With one million independent common variants in the human genome, the Bonferroni-corrected significance threshold becomes approximately 5 × 10⁻⁸, making it difficult to detect variants with small effect sizes without extremely large sample sizes [26] [25]. This fundamental trade-off between genomic coverage and statistical stringency has driven the formation of large international consortia to achieve sample sizes in the tens to hundreds of thousands.
Comparative Performance and Empirical Evidence
Replication Rates and Validation
The ultimate test of any genetic association study lies in its ability to produce replicable findings across independent samples. By this metric, GWAS has demonstrated clear advantages over traditional candidate gene approaches. Candidate gene studies have suffered from notoriously high rates of false positives and low replication rates across many fields, including psychiatry [25] [24]. Many initially promising candidate gene associations have failed to replicate in larger, more rigorous studies.
In contrast, GWAS-identified loci have shown remarkably consistent replication across diverse populations when sample sizes are sufficient. For instance, in a recent childhood B-cell acute lymphoblastic leukemia (B-ALL) GWAS involving 840 African American cases and 3,360 controls, multiple loci achieved genome-wide significance, with established trans-ancestral susceptibility regions at IKZF1 and ARID5B replicating previous findings [27]. Similarly, a GWAS meta-analysis of body weight traits in chickens identified 77 novel independent variants that were consistently associated across populations [28].
The table below illustrates quantitative comparisons from empirical studies:
Table 2: Empirical Performance Comparison from Published Studies
| Study Example | Approach | Sample Size | Significant Findings | Replication Success |
|---|---|---|---|---|
| B-ALL in African Americans [27] | GWAS | 840 cases, 3,360 controls | 9 genome-wide significant loci | 2 established loci replicated; 7 novel ancestry-specific loci |
| Chicken Body Weight Meta-analysis [28] | GWAS meta-analysis | 1,143 individuals across 3 populations | 77 novel variants | Consistent effects across populations |
| Chronic Post-Surgical Pain [29] | GWAS | 1,350 individuals | 77 SNPs in 24 loci | SNP-based heritability ~39% |
| Bovine Tuberculosis Susceptibility [26] | Candidate gene vs. GWAS simulation | 250-2,000 cases/controls | Candidate genes had higher power with known variants | GWAS struggled with weak effects in small samples |
Biological Insights and Novel Discoveries
Beyond mere replication, the true value of genetic studies lies in their ability to generate novel biological insights. GWAS has excelled in this domain, repeatedly identifying unsuspected biological pathways involved in complex diseases. The unbiased nature of GWAS has revealed that the majority of disease-associated variants reside in noncoding regions of the genome, suggesting they influence gene regulation rather than protein structure [25] [30] [24].
For example, GWAS of psychiatric disorders have identified numerous risk variants in genomic regions with no previously known connection to neurobiology, opening entirely new avenues for investigation [25]. Similarly, in sickle cell disease, GWAS have identified novel fetal hemoglobin-associated variants at loci including ASB3, BACH2, PFAS, ZBTB7A, and KLF1, which were subsequently replicated and shown to have functional effects in erythroid cells [31].
Candidate gene studies, while limited in discovery potential, can provide deep mechanistic insights when focused on truly causal genes. The key distinction is that GWAS is more reliable for identifying which genes warrant such intensive investigation.
Experimental Protocols and Methodological Frameworks
GWAS Workflow and Functional Validation
Modern GWAS follows a structured pipeline from genotyping to functional validation. The standard workflow begins with sample collection and genotyping using microarray platforms covering hundreds of thousands to millions of SNPs. After quality control and imputation to increase variant coverage, association analysis tests each variant for statistical association with the phenotype of interest [27] [28].
Significant associations undergo replication in independent samples to confirm validity, followed by fine-mapping to identify likely causal variants within associated regions [30]. The final and most challenging step involves functional validation using experimental methods to demonstrate biological mechanisms.
Figure 1: Standard GWAS Workflow from Sample Collection to Functional Validation
Functional Validation of GWAS Findings
Once GWAS identifies associated loci, determining their biological mechanisms requires sophisticated experimental approaches. Genome editing technologies, particularly CRISPR-based systems, have revolutionized this process by enabling precise modification of putative causal variants in relevant cell models [30].
A typical functional validation pipeline involves:
- Priority setting using functional genomic annotations (chromatin accessibility, TF binding sites)
- Regulatory element mapping through chromatin conformation capture (3C, Hi-C)
- Expression quantitative trait loci (eQTL) analysis to connect variants to target genes
- Genome editing in disease-relevant cell types
- Phenotypic assays to measure effects on gene expression and cellular functions [30]
For noncoding variants, protein binding assays including ChIP-Seq and electrophoretic mobility shift assays (EMSAs) can determine whether variants alter transcription factor binding affinities [30]. High-throughput approaches like SNP-seq enable unbiased identification of functional SNPs that allelically modulate regulatory protein binding [30].
Research Reagent Solutions and Experimental Tools
Success in both candidate gene and GWAS research depends on appropriate selection of research reagents and experimental platforms. The table below details essential tools and their applications:
Table 3: Essential Research Reagents and Experimental Platforms
| Reagent/Platform | Primary Function | Application Context | Key Considerations |
|---|---|---|---|
| SNP Microarrays | Genome-wide variant genotyping | GWAS discovery phase | Density (300K-5M variants), population-specific content |
| Whole Genome Sequencing | Comprehensive variant discovery | GWAS fine-mapping, rare variants | Coverage depth, structural variant detection |
| CRISPR-Cas9 Systems | Precise genome editing | Functional validation of causal variants | Delivery efficiency, off-target effects |
| ChIP-Seq Kits | Genome-wide protein-DNA interaction mapping | TF binding disruption by noncoding variants | Antibody specificity, cell type applicability |
| Reporter Gene Assays | Regulatory element activity measurement | Functional testing of putative enhancers | Minimal promoter choice, normalization controls |
| eQTL/SQTL Resources | Variant-gene expression association | Target gene prioritization | Cell type relevance, sample size |
Implications for Drug Development and Precision Medicine
The paradigm shift from candidate genes to GWAS has profound implications for therapeutic development. GWAS discoveries have increasingly revealed novel therapeutic targets in unsuspected biological pathways, expanding the potential intervention landscape for complex diseases [30]. The statistical robustness of well-powered GWAS findings provides greater confidence in investing in target validation and drug development programs.
Furthermore, GWAS findings enable polygenic risk scores that aggregate the effects of many variants to stratify individuals by disease risk. For instance, in the African American B-ALL study, children in the top polygenic risk score decile had a 7.9-fold greater odds of disease compared to those with median risk or lower [27]. Such risk prediction tools hold promise for targeting preventive interventions to high-risk subgroups.
However, challenges remain in translating GWAS findings to clinical applications. Most associated variants reside in noncoding regions with unclear functional consequences, and determining their target genes and mechanisms requires substantial additional investigation [30]. Additionally, the majority of GWAS have been conducted in European ancestry populations, limiting the translatability of findings across diverse populations—a concern that recent efforts have begun to address through more inclusive sampling [27].
The evolution from candidate gene to GWAS approaches represents genuine scientific progress in understanding complex trait genetics. Rather than completely replacing candidate gene research, GWAS has redefined its role—from initial discovery to focused mechanistic follow-up of robust genetic associations. The most productive research strategy now leverages the complementary strengths of both approaches: using GWAS for unbiased discovery of authentic genetic associations, then applying candidate gene-style intensive investigation to validate mechanisms and therapeutic implications [32] [30].
As genomic technologies continue advancing, this integrated approach will likely further evolve to include whole genome sequencing, multi-omics integration, and even larger-scale collaboration. The paradigm shift chronicled here has fundamentally improved the rigor, reliability, and biological insights from human genetics research, with lasting benefits for understanding disease mechanisms and developing targeted interventions.
Dopamine and serotonin represent two pivotal monoamine neurotransmitter systems that regulate a vast array of brain functions, including motivation, reward processing, mood stability, cognition, and motor control. The genetic architecture underlying these systems has become a primary focus in neuropsychiatric research, particularly for replication studies seeking to validate candidate genes across populations. Understanding the specific genes that encode synthesis enzymes, receptors, transporters, and metabolic components for these neurotransmitters provides crucial insights into individual differences in behavior, treatment response, and disease susceptibility [33] [34]. This guide systematically compares the key genetic elements of dopamine and serotonin pathways, summarizes experimental approaches for their investigation, and provides resources to facilitate rigorous replication studies in this domain.
Comparative Genetics of Dopamine and Serotonin Systems
Core Genetic Components of Major Neurotransmitter Pathways
Table 1: Key Gene Families in Dopamine and Serotonin Pathways
| System | Gene Category | Representative Genes | Protein Function | Chromosomal Location |
|---|---|---|---|---|
| Dopamine | Receptors | DRD1, DRD2, DRD3, DRD4, DRD5 | Dopamine receptor subtypes | Chromosome 5 (DRD1), 11 (DRD2), 3 (DRD3), 4 (DRD5) [33] |
| Synthesis & Metabolism | DDC, DBH, COMT | Dopamine synthesis (DDC), conversion to norepinephrine (DBH), degradation (COMT) | Chromosome 7 (DDC), 9 (DBH), 22 (COMT) [33] | |
| Transport | DAT1/SLC6A3, VMAT1/SLC18A1, VMAT2/SLC18A2 | Dopamine reuptake (DAT1), vesicular packaging (VMAT1/2) | Chromosome 5 (DAT1), 8 (VMAT1), 10 (VMAT2) [33] | |
| Serotonin | Receptors | HTR1A, HTR1B, HTR2A, HTR2C, HTR3A, HTR4, HTR5A, HTR6, HTR7 | Serotonin receptor subtypes | Multiple chromosomes [34] |
| Synthesis & Metabolism | TPH1, TPH2, DDC, MAOA | Serotonin synthesis (TPH1/2), conversion from 5-HTP (DDC), degradation (MAOA) | Chromosome 11 (TPH1), 12 (TPH2), 7 (DDC), X (MAOA) [34] | |
| Transport | SLC6A4/SERT | Serotonin reuptake | Chromosome 17 [35] |
Functionally Significant Polymorphisms and Associated Phenotypes
Table 2: Key Polymorphisms in Dopamine and Serotonin Genes and Their Research Implications
| Gene | Polymorphism | Functional Impact | Associated Phenotypes/Responses |
|---|---|---|---|
| COMT | rs4680 (Val158Met) | Met allele reduces enzyme activity by 4-fold, increasing synaptic dopamine [36] | Met/Met genotype linked to lower cooperation expectations in social dilemmas; influences motor learning response to rewards [37] [36] |
| DRD4 | 48-bp VNTR in exon III | 7-repeat allele associated with reduced receptor sensitivity [36] | Linked to reduced altruism, higher impulsivity, and aggression risk [36] |
| DAT1 | VNTR at 3'UTR | 9- and 10-repeat alleles most common; affects gene expression [35] | Associated with anxiety and stress sensitivity; epigenetic regulation under stress [35] |
| SLC6A4 | 5-HTTLPR (S/L alleles) | S allele reduces transcriptional efficiency; L allele increases serotonin reuptake [36] [35] | S allele: anxiety-related traits; L/L genotype: lower contributions in cooperative games without punishment [36] |
| HTR1B | rs13212041 (T/C) | T allele reduces gene expression via microRNA interaction [36] | T/T genotype associated with lower expectations of antisocial punishment [36] |
| Polygenic Score | Combined dopamine gene score | Summarizes cumulative genetic effects on dopamine transmission [37] | Predicts motor learning performance: low scores benefit from rewards, high scores learn better without rewards [37] |
Experimental Methodologies for Pathway Gene Investigation
Genetic Association Protocol: Dopamine-Related Motor Learning
Objective: To assess how individual variations in dopamine-related genes affect motor sequence learning with and without monetary rewards in children and young adults with and without cerebral palsy [37].
Participants: Inclusion of subjects aged 5-25 years with cerebral palsy and healthy volunteers, excluding those on medications affecting dopamine transmission (e.g., levodopa, methylphenidate) [37].
Genetic Analysis:
- Blood collection for genetic analysis of dopamine-related genes (DRD1-5, DAT1, COMT, DBH, DDC)
- Calculation of individual polygenic dopamine gene scores based on methodology by Pearson-Fuhrhop et al. [37]
- Stratification of participants into high and low gene score groups
Behavioral Task:
- Administration of serial reaction time task (SRTT) under two conditions:
- Unrewarded condition: Baseline performance assessment
- Rewarded condition: Monetary rewards for correct/rapid responses
- Measurement of learning rate as reduction in reaction times across sequence blocks
Statistical Analysis:
- Mixed-model ANOVA with gene score group as between-subject factor and reward condition as within-subject factor
- Post-hoc testing of gene score × condition interaction effects on learning rates [37]
Epigenetic Regulation Protocol: Transporter Gene Methylation in Stress
Objective: To evaluate DAT1 and SERT gene regulation through DNA methylation analysis in university students under perceived stress [35].
Participant Characterization:
- Administration of Highly Sensitive Person (HSP) scale (12-item) and Perceived Stress Scale (PSS-10)
- Stratification into low, medium, and high cumulative risk groups based on HSP × PSS interactions [35]
Sample Collection and Processing:
- Saliva collection (2 mL) after 2-hour fasting from food, drink (except water), smoking, or tooth brushing
- Genomic DNA extraction using salting-out method
- DNA methylation analysis at specific CpG sites in DAT1 5'UTR and SERT promoter regions [35]
Molecular Analysis:
- VNTR genotyping for DAT1 3'UTR (9- and 10-repeat alleles)
- Methylation-specific analysis of identified CpG sites
- Expression analysis of regulatory microRNAs (miR-491 for DAT1, miR-135 for SERT) [35]
Data Interpretation:
- Correlation of methylation patterns with stress sensitivity phenotypes
- Assessment of genotype × methylation interactions on stress vulnerability [35]
Signaling Pathway Architecture
The diagram above illustrates the parallel organization of dopamine and serotonin systems, highlighting shared elements like the AADC (DDC) enzyme while emphasizing system-specific components. Notably, the serotonin system originates from the essential amino acid tryptophan, while dopamine synthesis begins with tyrosine. Both systems employ similar regulatory mechanisms including transporter-mediated reuptake (SERT for serotonin, DAT for dopamine) and enzymatic degradation (MAOA for serotonin, COMT for dopamine), with the dopamine system additionally feeding into norepinephrine synthesis via DBH [33] [34].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Dopamine and Serotonin Gene Studies
| Reagent/Category | Specific Examples | Research Application | Function in Investigation |
|---|---|---|---|
| Genotyping Assays | TaqMan SNP Genotyping, VNTR analysis | Polymorphism screening (5-HTTLPR, COMT Val158Met, DAT1 VNTR) | Determines genetic variants of interest in candidate genes [37] [35] |
| DNA Methylation Kits | Bisulfite conversion kits, Methylation-specific PCR | Epigenetic regulation studies | Quantifies DNA methylation at promoter/regulatory regions of transporter genes [35] |
| RNA-Seq Platforms | Illumina, Thermo Fisher | Transcriptome profiling | Measures gene expression changes in response to experimental conditions [38] |
| Behavioral Task Software | Serial Reaction Time Task (SRTT), Weather Prediction Task (WPT), Public Goods Game | Cognitive and motor learning assessment | Quantifies behavioral phenotypes associated with genetic variants [37] [36] |
| LC-MS/MS Systems | HPLC with electrochemical or mass spectrometry detection | Neurotransmitter and metabolite quantification | Measures serotonin, dopamine, and metabolites (5-HIAA, DOPAC) [39] |
| microRNA Analysis Kits | miRNA extraction, qRT-PCR assays | Epigenetic regulation studies | Evaluates expression of miRNAs regulating transporter genes (miR-132, miR-491, miR-135) [35] |
Research Implications and Future Directions
The comparative analysis of dopamine and serotonin pathway genes reveals distinctive genetic architectures with important implications for replication studies. The dopamine system features a more diverse receptor family (DRD1-DRD5) distributed across multiple chromosomes, while serotonin signaling involves a more extensive receptor repertoire (HTR1-HTR7) with diverse signaling properties [33] [34]. Methodologically, comprehensive assessment requires integrated approaches spanning genetics (SNPs, VNTRs), epigenetics (DNA methylation, miRNA regulation), and precise behavioral phenotyping.
Future research directions should prioritize:
- Developing standardized polygenic scoring methods for cumulative genetic effects [37]
- Establishing best practices for epigenetic studies in accessible tissues (e.g., saliva) with brain relevance [35]
- Validating candidate genes across diverse populations and diagnostic categories
- Integrating multi-omics approaches to understand gene × environment interactions in neurotransmitter system regulation
These approaches will strengthen replication studies and facilitate the translation of genetic findings into personalized therapeutic strategies targeting dopamine and serotonin systems in neuropsychiatric disorders.
A Step-by-Step Protocol for Designing and Executing a Robust Replication Study
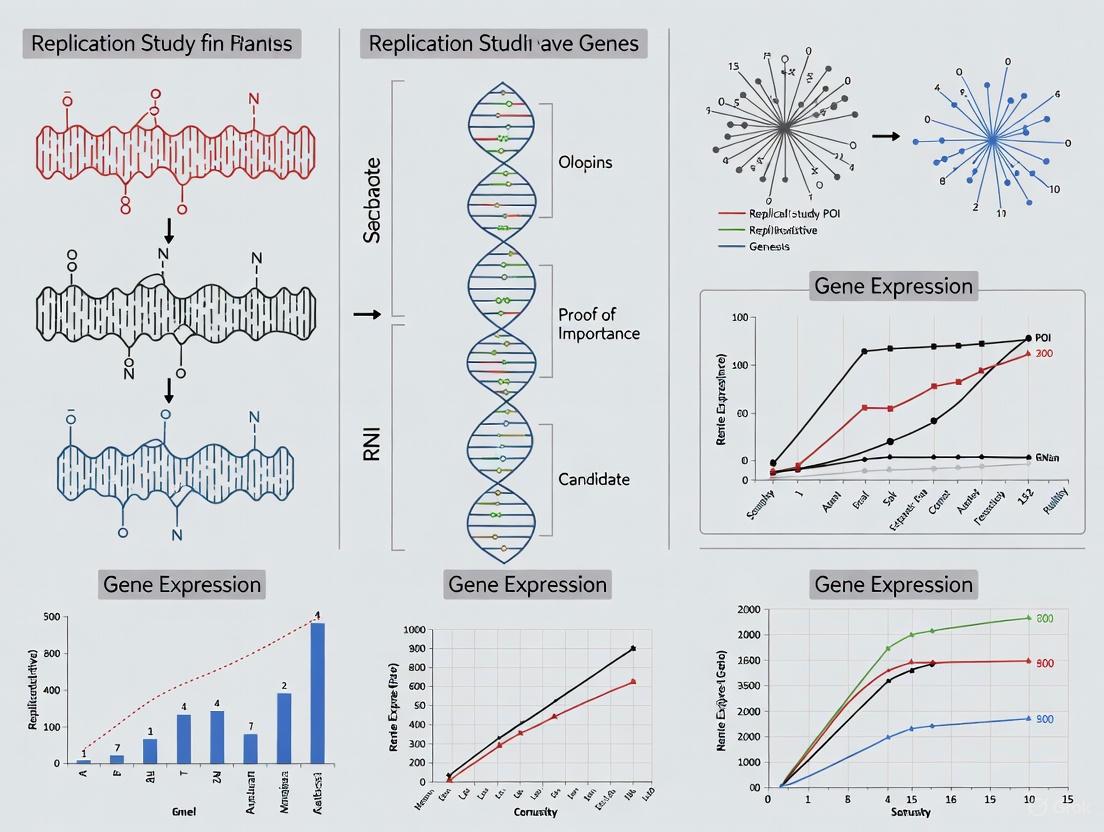
In the field of premature ovarian insufficiency (POI) research, the identification of candidate genes has accelerated dramatically, with over 90 genes now linked to this condition that affects 1-3.7% of women under 40 [40] [41]. This rapid expansion of genetic discoveries creates a critical bottleneck: determining which findings represent robust, reliable biological relationships worthy of further investigation and clinical translation. The selection of appropriate replication targets has thus become a fundamental challenge in the transition from initial discovery to validated knowledge. This guide provides a systematic framework for identifying the most valuable POI candidate genes for replication studies, enabling researchers to prioritize limited resources toward verifying the most promising genetic associations.
The POI Genetic Landscape: Quantifying Current Knowledge
POI represents a genetically heterogeneous condition, with established genetic causes accounting for approximately 20-25% of cases [42] [40]. Recent large-scale sequencing studies have significantly expanded our understanding of this genetic architecture. The table below summarizes the current genetic contribution to POI based on a 2023 study of 1,030 patients:
Table 1: Genetic Landscape of POI Based on Large-Scale Sequencing
| Genetic Category | Contribution to POI Cases | Key Genes and Examples |
|---|---|---|
| Overall Genetic Contribution | 23.5% (242/1030 cases) | Combinations of known and novel genes [4] |
| Known POI Genes | 18.7% (193/1030 cases) | 59 well-characterized genes [4] |
| Novel POI-Associated Genes | Additional 4.8% | 20 newly identified genes [4] |
| Chromosomal Abnormalities | 10-13% | Turner syndrome, X-structural variations [42] |
| Primary vs Secondary Amenorrhea | PA: 25.8% vs SA: 17.8% | Higher diagnostic yield in primary amenorrhea [4] |
The pathways implicated in POI pathogenesis are diverse, encompassing meiotic processes, DNA repair, mitochondrial function, and folliculogenesis. The diagram below illustrates the key biological pathways and their interrelationships in POI pathogenesis:
Core Criteria for Selecting Replication Targets
Assessing Value and Impact
The theoretical importance and potential clinical applicability of a genetic finding should be primary considerations in replication target selection. In POI research, value can be operationalized through specific criteria:
Clinical Impact: Genes associated with more severe phenotypic presentations typically warrant higher priority. For instance, variants in genes like
NR5A1andMCM9demonstrate relatively high prevalence among POI patients (1.1% each in recent studies) [4], suggesting broader clinical relevance.Biological Plausibility: Genes functioning in pathways critically important for ovarian development and function, particularly those with strong animal model evidence, represent stronger candidates. Meiosis and DNA repair genes constitute nearly half (48.7%) of genetically explained POI cases [4], highlighting this pathway's central importance.
Therapeutic Potential: Genes encoding potentially druggable targets or those that might inform clinical management decisions (e.g., cancer risk associations) should be prioritized. Recent research indicates that 37.4% of POI cases with genetic findings have implications for cancer susceptibility surveillance [41].
Evaluating Uncertainty and Robustness
Not all initial genetic associations are equally reliable. Assessing the degree of uncertainty surrounding a finding is crucial for replication prioritization:
Statistical Strength: Initial associations with modest statistical support require verification. Many POI gene discoveries originate from single-family studies without independent validation [41], representing high-uncertainty candidates.
Evidence Consistency: Genes supported by multiple independent case reports or functionally validated variants present stronger candidates. For example, the
EIF2B2gene showed the highest prevalence of pathogenic alleles in a recent study (0.8% of cases) due to a recurrent variant [4].Technical Considerations: Findings from studies with methodological limitations or those that have not employed optimal sequencing approaches (e.g., lack of trio-based sequencing to confirm de novo mutations) merit replication.
Considering Practical Feasibility
The practical aspects of replication studies significantly influence target selection:
Cohort Availability: Genes associated with more prevalent POI subtypes enable adequate sample collection. The higher diagnostic yield in primary amenorrhea (25.8%) versus secondary amenorrhea (17.8%) [4] suggests potentially easier recruitment for replication studies.
Variant Characterization: Well-documented variants with clear pathogenicity assessments are more straightforward to replicate. The 2023 Nature Medicine study established 195 pathogenic/likely pathogenic variants across 59 genes [4], providing a solid foundation for replication design.
Technical Infrastructure: Genes requiring specialized functional validation approaches (e.g., meiotic phenotyping) may present greater logistical challenges for replication.
Application to POI Candidate Genes
The following table applies the selection criteria to representative POI gene categories, providing a practical framework for replication prioritization:
Table 2: Replication Priority Assessment for POI Gene Categories
| Gene Category | Value/Impact | Uncertainty | Feasibility | Replication Priority |
|---|---|---|---|---|
| Meiosis/DNA Repair Genes (e.g., HFM1, MCM8, MCM9) | High (48.7% of solved cases) [4] | Medium (growing evidence) | Medium (requires specialized assays) | High |
| Syndromic Genes (e.g., AIRE, ATM) | Medium (multi-system involvement) | Low (well-established) | High (clinical features aid recruitment) | Medium |
| Novel Candidates (e.g., LGR4, PRDM1) | Unknown (pathway relevance) | High (limited evidence) | Variable | Research Question-Dependent |
| Mitochondrial Genes (e.g., AARS2, MRPS22) | Medium (22.3% of solved cases) [4] | Medium (emerging evidence) | Medium (requires functional studies) | Medium-High |
| FMR1 Premutation | High (well-established cause) | Low (extensively replicated) | High (standard testing available) | Low (already validated) |
Experimental Design for POI Gene Replication
Cohort Selection and Design Considerations
Successful replication requires careful attention to cohort composition and study design:
Sample Size Considerations: Large cohorts are essential for statistically meaningful replication. The 2023 study of 1,030 patients represents the current standard for sufficiently powered genetic studies in POI [4].
Phenotypic Stratification: Separating primary amenorrhea (PA) and secondary amenorrhea (SA) cases is crucial, as they demonstrate different genetic contributions (25.8% vs. 17.8% respectively) [4].
Population Considerations: Accounting for ethnic diversity is essential, as some genetic associations show population-specific effects.
Methodological Approaches
A tiered approach to replication provides comprehensive validation:
Technical Validation: Confirm initial variants using orthogonal methods (Sanger sequencing, etc.).
Independent Cohort Replication: Recruit new patient cohorts matching original study characteristics.
Functional Validation: Implement pathway-specific assays to confirm biological impact.
The diagram below illustrates a comprehensive replication workflow for POI candidate genes:
Essential Research Reagents and Solutions
The table below outlines key reagents and methodologies essential for conducting replication studies in POI genetics:
Table 3: Research Reagent Solutions for POI Gene Replication Studies
| Reagent/Methodology | Primary Function | Application in POI Research |
|---|---|---|
| Whole Exome Sequencing | Comprehensive coding variant detection | Initial gene discovery and variant identification [4] |
| Sanger Sequencing | Targeted variant confirmation | Orthogonal validation of putative pathogenic variants [4] |
| Array CGH | Copy number variation detection | Identification of chromosomal structural variants [42] |
| FSH/LH/AMH Assays | Hormonal profiling | Phenotypic characterization of POI patients [40] |
| T-clone/10x Genomics | Phase determination | Establishing cis/trans configuration of multiple variants [4] |
| Functional Assays | Pathogenicity assessment | MEIOSIN, CPEB1 validation for meiotic genes [4] |
Systematic selection of replication targets in POI research requires balanced consideration of multiple factors, with genes involved in DNA repair and meiotic pathways currently representing the highest-value candidates based on their substantial contribution to explained cases. As the field evolves toward personalized medicine approaches—where genetic diagnoses may inform cancer risk management (37.4% of cases) [41] or potential fertility interventions—the importance of robust, replicated genetic data becomes increasingly critical. By applying the structured framework presented in this guide, researchers can prioritize their replication efforts to maximize scientific yield and accelerate the translation of genetic discoveries to clinical applications in ovarian insufficiency.
In the field of genetic epidemiology, the "winner's curse" represents a critical statistical phenomenon that profoundly impacts the validation of disease-associated genetic variants. When a variant is identified as significant in an initial genomewide association study (GWAS), the estimated genetic effect size is often substantially inflated, particularly when the original study had low or moderate statistical power [43]. This overestimation occurs because, for a genuinely associated variant with modest effect, only those chance occurrences where the effect appears largest will reach statistical significance threshold [43]. This ascertainment bias has far-reaching implications, as it frequently leads to underpowered replication studies that fail to confirm genuine associations, ultimately wasting scientific resources and delaying discoveries [43] [44].
The challenge is particularly acute in complex genetic disorders like premature ovarian insufficiency (POI), where researchers must identify genuine genetic signals from a sea of candidates. POI affects approximately 1-2% of women before age 40 and represents a clinically heterogeneous condition with substantial knowledge gaps in its etiology [45] [46]. Nearly 70% of POI cases remain unexplained, making genetic discovery a paramount research priority [45]. In this context, properly addressing the winner's curse through rigorous power and sample size calculation becomes not merely a statistical formality, but a fundamental requirement for robust gene discovery and replication.
Statistical Foundations: Understanding Power, Sample Size, and Effect Size
The Interrelationship of Key Statistical Parameters
Statistical power, sample size, effect size, and significance thresholds form an interconnected framework that determines the validity of genetic association studies. Power, defined as the probability of correctly rejecting a false null hypothesis (1-β), is critically dependent on three factors: the chosen significance threshold (α), the true effect size of the variant, and the sample size available for analysis [44]. The delicate balance between Type I error (false positives) and Type II error (false negatives) must be carefully managed through appropriate experimental design [44].
The traditional convention in scientific research sets α at 0.05 and power at 0.8 (80%), though these values should be adjusted based on the specific research context [44]. For pilot studies with exploratory aims, α may be relaxed to 0.10 or 0.20, while in situations with severe consequences for false positives (such as drug development studies), α might be set at 0.001 or lower [44]. Understanding these relationships is essential for designing replication studies that can overcome the winner's curse.
Quantifying the Winner's Curse Effect
The magnitude of the winner's curse bias is inversely related to the power of the initial study. When power is high (e.g., >90%), most random samples from the true distribution will yield significant results, making the ascertainment bias minimal. However, when power is low, conditioning on significant association in the discovery phase creates substantial upward bias in effect size estimates [43]. Empirical evidence demonstrates this problem is widespread in genetic association studies. A meta-analysis of 301 studies across 25 putative disease loci found that 24 of the 25 initial reports showed higher odds ratios than subsequent replication studies [43].
This effect has direct consequences for POI research, where genetic variants often have modest effects and initial sample sizes may be limited. If replication studies are designed based on inflated effect sizes from initial discoveries, the sample sizes will be insufficient to detect the true, more modest effects, leading to failure in replication and potentially abandoning genuine associations [43].
Methodological Approaches for Bias Correction
Statistical Methods for Correcting Parameter Estimates
Several methodological approaches have been developed to correct for the winner's curse in genetic association studies. The maximum likelihood estimation (MLE) approach provides a framework for generating corrected estimates of penetrance and allele frequency parameters [43]. This method calculates the likelihood of the observed genotype counts conditional on having detected a significant association, effectively weighting the naive estimates by the power of the initial test [43].
The mathematical foundation of this approach can be represented as:
L(θ,φ) = Pr(D|θ,φ,S) / Pr(B|θ,φ,S)
Where D represents the observed genotype counts, B indicates a significant association, S denotes the sampling design, θ represents the penetrance parameters, and φ represents the genotype frequencies [43]. The denominator represents the statistical power to detect association, which acts as a correction factor, tilting the maximum likelihood toward more conservative effect sizes [43].
Alternative approaches include the two-stage method that randomly splits samples into discovery and estimation sets, though this approach produces estimates with higher standard errors [43]. Another method lowers the significance threshold in the initial test to increase power and reduce ascertainment bias, though this comes at the cost of increased false positives [43].
Sample Size Calculation for Replication Studies
Proper sample size calculation for replication studies must account for the corrected effect sizes rather than the naive estimates from the initial discovery. The required sample size depends on the specific genetic model (additive, dominant, recessive), minor allele frequency, and the corrected effect size [43] [44].
Table 1: Sample Size Calculation Formulas for Different Study Types
| Study Type | Formula | Parameters |
|---|---|---|
| Two proportions | (n = \frac{(Z{1-\alpha/2} + Z{1-\beta})^2 \times [p1(1-p1) + p2(1-p2)]}{(p1 - p2)^2}) | p₁, p₂ = event proportions in groups [44] |
| Two means | (n = \frac{(Z{1-\alpha/2} + Z{1-\beta})^2 \times 2\sigma^2}{d^2}) | σ = pooled standard deviation, d = difference of means [44] |
| Odds ratio | (n = \frac{(Z{1-\alpha/2} + Z{1-\beta})^2}{[ln(OR)]^2 \times \frac{1}{p1(1-p1)} + \frac{1}{p2(1-p2)}}) | OR = odds ratio, p₁, p₂ = event proportions [44] |
| Correlation | (n = \frac{(Z{1-\alpha/2} + Z{1-\beta})^2}{[0.5 \times ln(\frac{1+r}{1-r})]^2} + 3) | r = correlation coefficient [44] |
These calculations can be implemented using various statistical software packages and online calculators, though understanding their theoretical foundations remains essential for appropriate application [44].
Application to POI Candidate Gene Research
Current Genetic Research Landscape in POI
POI research has increasingly relied on advanced genetic technologies to identify causative variants. Recent studies utilize a combination of array comparative genomic hybridization (array-CGH) and next-generation sequencing (NGS) panels targeting genes known or suspected to be involved in ovarian function [45]. This approach has identified genetic anomalies in 57.1% (16/28) of idiopathic POI patients, with causal copy number variations found in 3.6% and causal single nucleotide variations/indels in 28.6% [45].
The complex genetic architecture of POI means that most individual variants have modest effects, making them particularly susceptible to the winner's curse. Family studies demonstrate the strong genetic component of POI, with familial forms identified in 12-31% of cases [45]. The emerging understanding of POI pathophysiology includes abnormalities in follicular pool establishment, accelerated follicular atresia, and alterations in primordial follicle recruitment [45].
Research Workflow in POI Genetic Studies
The standard research workflow in contemporary POI genetic studies incorporates multiple validation steps to ensure robust findings, though additional attention to power considerations is needed to fully address the winner's curse.
Experimental Protocols for POI Genetic Studies
Sample Collection and Diagnostic Criteria
Well-defined patient cohorts are fundamental to robust genetic studies of POI. Current research typically includes women meeting the following criteria: (i) age < 40 years; (ii) at least 4 months of oligo/amenorrhea; and (iii) elevated serum follicle-stimulating hormone (FSH) > 25 IU/L on two occasions至少4 weeks apart [46]. Exclusion criteria generally encompass other endocrine diseases, history of ovarian surgery, and recent hormone use (within 3 months) [46]. Control groups typically consist of women with infertility due to tubal factors but with normal menstrual cycles and basal sex hormone levels [46].
Genetic Analysis Techniques
Comprehensive genetic analysis in POI research involves multiple complementary approaches:
Array-CGH: Conducted using platforms such as SurePrint G3 Human CGH Microarray 4 × 180 K technology (Agilent Technologies), with data analysis using Feature Extraction and CytoGenomics software. This technique identifies copy number variations (CNVs) with minimum resolution of 60 kb [45].
Next-Generation Sequencing: Performed using capture-based targeted sequencing (e.g., SureSelect XT-HS) of gene panels encompassing 163 genes associated with ovarian function. Sequencing occurs on platforms such as Illumina NextSeq 550, with variant calling using Alissa Align&Call and interpretation with Alissa Interpret [45].
Third-Generation Sequencing: Emerging approaches utilizing Oxford Nanopore Technology (ONT) enable full-length transcript characterization, overcoming limitations of short-read sequencing and improving detection of structural variants [46].
Validation Methods
Independent validation of genetic findings employs several experimental approaches:
qRT-PCR: Used to confirm expression changes of candidate genes in independent sample sets. RNA is extracted from monocytes or granulosa cells, reverse transcribed, and quantified using SYBR Green chemistry with normalization to housekeeping genes like GAPDH [46].
Functional Assays: Include in vitro models to test the impact of genetic variants on protein function, pathway analysis, and biomarker development.
Table 2: Essential Research Reagents for POI Genetic Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| RNA Collection | PAXgene Blood RNA tubes (BD) | Standardized RNA preservation from peripheral blood [46] |
| RNA Extraction | QIAsymphony DNA midi kits (Qiagen) | Automated nucleic acid extraction [45] |
| Microarray | SurePrint G3 Human CGH Microarray 4×180K (Agilent) | CNV detection [45] |
| NGS Library Prep | SureSelect XT-HS (Agilent) | Target enrichment for sequencing [45] |
| Sequencing Platforms | Illumina NextSeq 550, Oxford Nanopore PromethION | DNA and RNA sequencing [45] [46] |
| Variant Interpretation | CytoGenomics, Alissa Interpret, STRING, Cytoscape | Bioinformatic analysis [45] [46] |
| Validation | SYBR Green qPCR Master Mix (ServiceBio) | Gene expression validation [46] |
Comparative Analysis of Statistical Approaches
Evaluating Correction Methods for Genetic Studies
Different statistical approaches for addressing the winner's curse offer distinct advantages and limitations in the context of POI research. The table below summarizes key methodologies and their applications:
Table 3: Comparison of Statistical Methods for Overcoming Winner's Curse
| Method | Key Principle | Advantages | Limitations | Relevance to POI |
|---|---|---|---|---|
| Maximum Likelihood Estimation [43] | Conditions estimates on significance in initial study | Provides point estimates and confidence regions; greatly reduces bias | Complex computation; requires specification of genetic model | High - suitable for well-defined genetic models in POI |
| Sample Splitting [43] | Randomly divides data into discovery and estimation sets | Simple implementation; minimal technical expertise | Reduced power in both phases; higher standard errors | Moderate - useful for large cohorts with sufficient samples |
| Significance Threshold Adjustment [43] | Lowers α in initial test to increase power | Reduces ascertainment bias through higher power | Increases false positives; requires more stringent final thresholds | Low - limited by typically small POI cohort sizes |
| Bootstrapping/Resampling | Empirical estimation of bias through resampling | Non-parametric; makes minimal assumptions | Computationally intensive; may underestimate extreme biases | Moderate - valuable for complex genetic models |
Power Considerations in POI Study Design
The implementation of appropriate power calculations requires careful consideration of POI-specific research challenges. Family-based designs offer increased power for detecting rare variants but require specialized recruitment strategies [45]. Case-control association studies are more feasible but require larger sample sizes to achieve adequate power for modest genetic effects [43] [44].
Recent research has identified several promising genetic biomarkers for POI through advanced methodologies, including COX5A, UQCRFS1, LCK, RPS2, and EIF5A, which demonstrate consistent expression trends in validation studies [46]. These findings highlight the importance of oxidative phosphorylation pathways and DNA damage repair mechanisms in POI pathophysiology [46]. The convergence of these biological pathways through independent genetic studies strengthens their validity and provides promising targets for therapeutic development.
Implementation Framework for POI Research
Integrated Workflow for Robust Genetic Discovery
Building on the statistical principles and methodological considerations discussed, the following pathway diagram illustrates an integrated approach to POI genetic research that systematically addresses the winner's curse throughout the research process:
Future Directions and Recommendations
As POI genetic research advances, several key considerations will enhance the field's ability to overcome the winner's curse and produce more reliable findings:
Collaborative Consortia: Given the relative rarity of POI and the modest effect sizes of most genetic variants, multi-center collaborations are essential to achieve sample sizes sufficient for well-powered discovery and replication [45] [46].
Standardized Phenotyping: Implementation of consistent diagnostic criteria across research centers will improve cohort homogeneity and enhance statistical power [45] [46].
Advanced Statistical Methods: Incorporation of machine learning approaches such as random forest and Boruta algorithms can enhance feature selection and identify genuine genetic signals amidst multiple candidates [46].
Multi-Omics Integration: Combining genomic data with transcriptomic, epigenomic, and proteomic information will provide complementary evidence for genetic associations and enhance biological validation [47] [46].
The continued development and application of robust statistical methods for power calculation and bias correction will ensure that POI genetic research produces increasingly reliable findings, ultimately advancing our understanding of this complex disorder and leading to improved diagnostic and therapeutic approaches.
The replication of genetic associations in Premature Ovarian Insufficiency (POI) research represents a critical challenge in the field of reproductive genetics. POI, characterized by the loss of ovarian function before age 40, affects 1-3.7% of women and poses a significant cause of female infertility [48] [4] [40]. Despite the identification of numerous candidate genes through advanced sequencing technologies, a substantial proportion of POI cases remain idiopathic, underscoring the complex and heterogeneous nature of this condition [40]. The replication crisis in POI genetics stems from inconsistent phenotype documentation, variable environmental context reporting, and methodological disparities across studies. This guide systematically addresses these challenges by providing standardized frameworks for phenotype-environment documentation and analytical protocols, aiming to enhance the reliability and reproducibility of POI candidate gene research for scientists, researchers, and drug development professionals.
Phenotype Documentation Standards
Core Diagnostic Criteria
Comprehensive phenotype documentation forms the foundation of replicable POI genetic research. The minimal diagnostic criteria established by professional societies provide a baseline, but replication requires deeper phenotypic characterization.
Table 1: Essential Phenotypic Elements for POI Genetic Studies
| Category | Essential Elements | Standardized Values | Reporting Requirements |
|---|---|---|---|
| Menstrual Status | Primary vs secondary amenorrhea; Duration | Months of amenorrhea; Age at onset | Required for all participants |
| Hormonal Profile | FSH, LH, AMH, Estradiol | IU/L for FSH/LH; ng/mL for AMH | Two measurements ≥4 weeks apart |
| Ovarian Morphology | Antral follicle count; Ovarian volume | Count; mL | Transvaginal ultrasound preferred |
| Age Parameters | Age at diagnosis; Age at amenorrhea | Years | Exact ages required |
Beyond these core elements, comprehensive phenotyping should capture the clinical heterogeneity of POI. Primary amenorrhea (PA) versus secondary amenorrhea (SA) represents a crucial distinction, as studies demonstrate different genetic contribution profiles between these presentations [4]. Specifically, cases with PA show a higher prevalence of biallelic and multi-het pathogenic variants compared to SA cases (8.3% vs 3.1%), suggesting more substantial genetic contributions in PA [4]. Additional parameters should include response to ovarian stimulation (where available), family history of POI or other infertility conditions, and associated autoimmune or metabolic features.
Phenotypic Subcategorization Strategy
Stratifying POI cases based on phenotypic features enhances genetic discovery and replication potential. Research indicates distinct genetic architectures across POI subtypes:
- Syndromic vs Isolated POI: Approximately 8.5% of POI cases represent the only visible manifestation of a multi-organ genetic disorder [48]. Documenting extra-ovarian features is essential for identifying syndromic forms.
- Ovarian Reserve Spectrum: While the diagnosis requires elevated FSH and amenorrhea, capturing the full spectrum of ovarian reserve through AMH values and antral follicle count enables stratification for genetic analysis [49].
- Family History Patterns: Documenting the presence of POI in first-degree relatives (mothers, sisters) or extended family members helps identify potentially monogenic versus polygenic forms [49].
Environmental Context Documentation
Environmental Modifiers and Exposures
Environmental factors interact with genetic predispositions to influence POI presentation and progression. Comprehensive documentation of these factors is essential for controlling confounding variables in replication studies.
Table 2: Environmental Exposures and Modifiers Documentation Protocol
| Exposure Category | Specific Elements | Measurement Method | Timing Assessment |
|---|---|---|---|
| Iatrogenic Exposures | Chemotherapy agents; Radiation dose/site; Surgical history | Medical record review; Self-report with verification | Pre-POI onset timeline |
| Autoimmune Context | Autoimmune disorders; Autoantibody profiles | Medical diagnosis; Laboratory results | At POI diagnosis |
| Lifestyle Factors | Smoking history; BMI trajectory; Occupational exposures | Pack-years; Longitudinal BMI; Exposure questionnaires | Pre- and post-diagnosis |
| Medication History | Hormonal treatments; Immunosuppressants | Medication name, duration, dose | Prior to amenorrhea onset |
Environmental documentation should specifically capture exposures preceding POI onset, as these may trigger ovarian insufficiency in genetically susceptible individuals. The exclusion criteria implemented in major studies typically include iatrogenic causes (chemotherapy, radiation, ovarian surgery), confirmed autoimmune etiologies, and active endocrine pathologies [49] [4]. Precise documentation enables appropriate stratification in genetic analyses.
Gene-Environment Interaction Framework
The environment-to-phenotype mapping provides a theoretical framework for understanding how environmental cues interact with genetic predispositions to influence phenotypic outcomes [50]. In POI, this mapping can manifest through several adaptive strategies:
- Unvarying Strategy: Genetic predisposition consistently leads to POI regardless of environmental context
- Tracking Strategy: Environmental cues (e.g., chemical exposures) trigger POI in genetically susceptible individuals
- Bet-Hedging Strategy: Stochastic environmental interactions explain variable penetrance in genetic carriers
Documenting environmental contexts enables researchers to model these interaction patterns and improve replication across diverse cohorts.
Genetic Analysis Methodologies
Candidate Gene Validation Protocols
The validation of POI candidate genes requires rigorous variant identification, filtering, and annotation protocols. Whole-exome sequencing (WES) has emerged as a powerful tool for identifying causative genes in both familial and sporadic POI cases [49] [48] [4].
Table 3: Variant Filtering and Annotation Criteria in POI Studies
| Filtering Step | Parameters | Typical Values | Purpose |
|---|---|---|---|
| Quality Filtering | Read depth; Mapping quality; Genotype quality | ≥10× coverage; QScore ≥30 | Remove technical artifacts |
| Frequency Filtering | Minor allele frequency (MAF) | <0.01 in population databases (gnomAD) | Exclude common polymorphisms |
| Variant Impact | Predicted functional consequence | LoF, missense, splice-site | Prioritize potentially damaging variants |
| Segregation Analysis | Familial co-segregation | Present in all affected family members | Support causality in familial cases |
| Pathogenicity Prediction | ACMG guidelines; In silico tools | CADD ≥20; Multiple supporting algorithms | Classify pathogenic/likely pathogenic variants |
The application of these filtering strategies in a study of 7 POI families identified 23 potentially damaging variants in 22 genes, all heterozygous, with five families carrying variants in multiple genes [49]. This highlights the potential polygenic etiology in some POI cases.
Case-Control Association Frameworks
Robust case-control designs are essential for establishing statistical evidence for novel POI gene associations. The largest WES study to date compared 1,030 POI cases with 5,000 controls, identifying 20 novel POI-associated genes through a significantly higher burden of loss-of-function variants [4]. Key considerations for case-control studies include:
- Control Selection: Population-matched controls without POI diagnosis, with normal ovarian function confirmed where possible
- Power Considerations: Large sample sizes needed to achieve genome-wide significance given multiple testing burden
- Phenotype Matching: Careful matching or stratification based on POI subtype (PA vs SA) and family history
This experimental design enabled the identification of novel genes involved in gonadogenesis (LGR4, PRDM1), meiosis (CPEB1, KASH5, MCMDC2), and folliculogenesis (ALOX12, BMP6, ZP3) [4].
Experimental Replication Workflow
The following diagram illustrates the comprehensive workflow for replicating POI candidate gene associations, integrating phenotypic documentation, genetic analysis, and functional validation:
Integrated Data Analysis Approaches
Multi-Omics Correlation Frameworks
Advanced replication studies increasingly integrate multiple data types to strengthen candidate gene validation. Transcriptomic analyses of granulosa cells and endometrial tissues have identified hub genes like CENPW, ENTPD3, FOXM1, GNAQ, LYPLA1, and PLA2G4A that may connect POI with other reproductive conditions like recurrent spontaneous abortion [51]. Integration strategies include:
- Co-expression Analysis: Identifying networks of genes differentially expressed in POI tissues
- Pathway Enrichment: Mapping candidate genes to biological pathways (e.g., oxidative phosphorylation, ribosome processes, steroid biosynthesis)
- Regulatory Network Mapping: Constructing transcription factor and miRNA-gene regulatory networks
These approaches move beyond single-gene associations to pathway-level validation, enhancing biological plausibility and replication potential.
Functional Annotation Pipelines
Candidate genes require functional annotation to interpret their potential roles in ovarian biology. Key annotation resources include:
- Expression Databases: GTEx portal for tissue-specific expression patterns
- Protein-Protein Interactions: STRING database for interaction networks
- Model Organism Phenotypes: Mouse Genome Informatics for reproductive phenotypes
- Chromosomal Interaction Data: Hi-C data for regulatory element connections
For example, genes implicated in DNA repair and meiosis (37.4% of cases in one large study) often represent tumor/cancer susceptibility genes as well, necessitating lifelong monitoring for associated comorbidities [48].
Research Reagent Solutions
Table 4: Essential Research Reagents and Platforms for POI Genetic Studies
| Reagent Category | Specific Products/Platforms | Application in POI Research | Key Considerations |
|---|---|---|---|
| Sequencing Platforms | Illumina NextSeq; Trusight One Panel | Whole-exome sequencing; Targeted gene panels | Coverage depth >98% at 10×; Mean read depth 100-180× [49] |
| Variant Annotation | Variant Interpreter; ANNOVAR; CADD | Functional prediction of identified variants | Integrate multiple algorithms (SIFT, PolyPhen-2) [49] |
| CNV Detection | DNAcopy package; Read-depth based methods | Identification of copy number variations | Circular binary segmentation algorithm implementation [48] |
| Expression Analysis | RNA-seq; qRT-PCR validation | Transcriptomic profiling in ovarian tissues | Validation in granulosa cells and endometrial tissues [51] |
| Functional Validation | CRISPR/Cas9; Immunoassays; Mitomycin C assay | Mechanistic studies of gene function | Chromosomal breakage analysis for DNA repair genes [48] |
The replication of POI candidate gene research demands meticulous attention to phenotypic documentation, environmental context, and standardized analytical frameworks. By implementing the comprehensive protocols outlined in this guide—including detailed phenotype characterization, rigorous variant filtering, case-control association designs, and functional validation pathways—researchers can significantly enhance the reliability and reproducibility of genetic discoveries in POI. The integration of multi-omics data and pathway-level analyses further strengthens the biological plausibility of candidate genes. As the field advances, these standardized approaches will facilitate the translation of genetic findings into improved diagnostic capabilities, personalized therapeutic strategies, and ultimately, better outcomes for women affected by premature ovarian insufficiency.
The replication crisis has underscored a critical flaw in empirical research, revealing that many highly influential findings are less robust than they initially appear [52]. This crisis of confidence has fundamentally undermined the trust researchers place in published literature, prompting a methodological revolution focused on improving research quality and restoring scientific credibility [53]. Within this context, preregistration has emerged as a powerful corrective measure—a process where researchers articulate their research plans including rationale, hypotheses, design, analysis, and sampling plans before collecting and analyzing data [53]. For replication studies of candidate genes research, where false positives have been particularly problematic, preregistration offers a systematic approach to distinguish confirmatory hypothesis testing from exploratory analysis, thereby preserving the diagnosticity of statistical inferences [54].
The theoretical foundation of preregistration rests on counteracting well-documented cognitive biases and questionable research practices (QRPs). Hindsight bias (the "I-knew-it-all-along" effect) causes researchers to see outcomes as more predictable after observing data, leading them to mistake postdictions for predictions [54]. Similarly, practices like p-hacking (manipulating data collection or analysis until achieving statistical significance) and HARKing (hypothesizing after results are known) artificially inflate the apparent strength and reliability of findings [53]. Preregistration addresses these issues by creating a time-stamped, read-only plan that locks in analytical decisions before data observation, thus providing a transparent record of what was originally planned versus what was ultimately discovered [55].
Understanding Preregistration: Concepts and Mechanisms
Core Principles and Terminology
Preregistration operates on a fundamental distinction between confirmatory and exploratory research. Confirmatory research tests specific, predetermined hypotheses with the goal of minimizing false positives (Type I errors), whereas exploratory research seeks potential relationships, effects, or differences with the goal of minimizing false negatives (Type II errors) [55]. This distinction is crucial because statistical inference tools, including null hypothesis significance testing (NHST), are designed for testing predictions, not generating them [54].
Several key terms are essential for understanding preregistration:
- Preregistration: The practice of submitting a time-stamped, read-only study plan to a public repository before beginning data collection or analysis [56]. The term emphasizes that submission occurs before the study begins.
- Registration: Originating from clinical research, this refers to submitting a study plan to a public registry, with "prospective" registration defined as occurring prior to enrolling the first patient [56].
- Pre-analysis plan: A term originating in economics referring to the data analysis procedures that will be used, created prior to accessing or collecting data [56].
- Registered Reports: An extension of preregistration where the study protocol undergoes peer review before data collection, with journals providing in-principle acceptance regardless of eventual results [57].
The Theoretical Mechanism: How Preregistration Reduces Bias
Preregistration functions as a safeguard against bias through several interconnected mechanisms. By requiring researchers to specify their hypotheses, methodology, and analysis plan before data collection, it creates a verifiable distinction between planned confirmatory tests and unplanned exploratory analyses [54]. This distinction is critical because it prevents researchers from mistakenly presenting postdictions (explanations generated after seeing data) as predictions (hypotheses generated before seeing data)—a confusion that leads to overconfidence in research findings [54].
The process specifically addresses what Gelman and Loken term the "garden of forking paths"—the vast number of analytical choices available to researchers [54]. When these choices are made during analysis, observing the data inevitably influences which paths are selected, rendering statistical inferences like p-values uninterpretable. Preregistration avoids this problem by specifying the analytical pipeline before data observation, thus preserving the diagnosticity of statistical tests [54].
For replication studies, this mechanism is particularly valuable. Preregistration ensures that replication attempts maintain methodological fidelity to original studies while pre-specifying criteria for determining whether results support or contradict original findings [56]. This prevents both unintentional methodological drift and conscious or unconscious manipulation of criteria to achieve desired outcomes.
Platform Comparison: OSF Versus Alternatives
Evaluation Framework and Core Criteria
When selecting a preregistration platform, researchers must consider both minimum essential features and additional functionality that enhances usability. Based on comparative analysis of popular platforms serving broad research fields, three criteria constitute the bare minimum for a valid preregistration: (1) an immutable timestamp to verify when the plan was created; (2) a public registry for accessibility and transparency; and (3) persistence to ensure the record remains permanently available [58] [59]. Additional valuable features include template variety, integration capabilities, and flexibility for different research types.
Table 1: Platform Comparison Based on Essential Preregistration Criteria
| Platform | Timestamp | Registry | Persistence | Additional Features |
|---|---|---|---|---|
| OSF | Yes | Yes | Yes | Multiple templates, project integration, embargo options |
| AsPredicted | Limited [58] | No [58] | Limited [58] | Streamlined process, minimal guidance |
| Zenodo | Yes | Yes | Yes | DOI assignment, general-purpose repository |
| GitHub | Limited [58] | No [58] | Limited [58] | Version control, collaboration features |
Table 2: Specialized Platform Features and Research Applications
| Platform | Template Variety | Best For | Limitations |
|---|---|---|---|
| OSF | 11+ templates including quantitative, qualitative, and secondary data analysis [56] | First-time users, multidisciplinary teams, complex designs | Can be overwhelming for simple studies |
| AsPredicted | Single streamlined template | Simple confirmatory studies, quick registration | Limited guidance, fails basic criteria [58] |
| ClinicalTrials.gov | Clinical trial specific | FDA-regulated clinical trials | Limited to specific study types [59] |
| PROSPERO | Systematic review specific | Systematic reviews and meta-analyses | Limited to specific study types [59] |
In-Depth Platform Analysis
Open Science Framework (OSF) emerges as the most fully-featured platform, meeting all three basic criteria while offering extensive additional functionality. OSF provides researchers with multiple workflow options—they can create a standalone registration or register an existing OSF project to freeze it at a specific point in time [56]. The platform offers numerous discipline-specific templates, including the comprehensive OSF Preregistration template, Preregistration in Social Psychology, Qualitative Preregistration, and specialized templates for secondary data analysis and replication studies [56]. This template diversity makes OSF particularly valuable for complex replication studies in candidate genes research, where precise methodological specification is crucial.
OSF also addresses common researcher concerns through thoughtful features. For those worried about idea scooping, OSF allows registrations to be kept private for up to four years [60]. The platform also accommodates necessary changes through registration updates or "Transparent Changes" documents, recognizing that preregistrations should be "a plan, not a prison" [60] [57].
AsPredicted offers a simplified alternative with a minimalist approach, asking only essential questions with minimal guidance [56]. While this streamlined process may appeal to those conducting straightforward confirmatory studies, the platform's failure to meet all three basic preregistration criteria limits its reliability for establishing research credibility [58].
Zenodo, a general-purpose repository, meets the basic preregistration criteria and assigns Digital Object Identifiers (DOIs) to registered plans [58]. However, it lacks the discipline-specific templates and integrated research workflow features that make OSF particularly useful for managing the entire research lifecycle.
Specialized registries like ClinicalTrials.gov for clinical trials and PROSPERO for systematic reviews offer domain-specific functionality but are limited to particular research types [59], making them unsuitable for most candidate gene replication studies.
Experimental Evidence: Quantifying Preregistration Benefits
Impact on Effect Size Estimation and Reproducibility
Empirical research demonstrates that preregistration substantively improves research credibility by addressing systemic biases in effect size estimation. In a theoretical and empirical analysis, researchers found that in settings with selective reporting and low statistical power, effect sizes become highly inflated, leading to low reproducibility [61]. Preregistration was shown to mitigate this inflation bias by increasing the share of researchers who adhere to predefined analytical plans ("frequentist researchers"), thus producing more accurate effect size estimates [61]. Numerical applications of the statistical model indicated that this inflation bias could be very large in practice, with available empirical evidence supporting the central assumptions of the model [61].
This effect size inflation has direct consequences for replication planning. When original studies report inflated effect sizes, replication studies designed based on those estimates are underpowered to detect the true (typically smaller) effects [61]. Preregistration therefore improves not only the credibility of original findings but also the efficiency and diagnostic value of replication attempts—a critical consideration for candidate genes research where replication failures have been particularly common.
Effect on Trust and Perceived Credibility
The relationship between preregistration and perceived trustworthiness represents another important dimension of its impact. A registered report study explicitly investigated whether preregistration increases the trust that fellow researchers place in reported outcomes [53]. While the study ultimately reported ambiguous Bayes factors and methodological challenges that limited definitive conclusions, the very existence of this research demonstrates the central role that trust plays in the scientific discourse around preregistration [53].
The theoretical basis for expecting preregistration to enhance trust is robust. By making the research process more transparent and minimizing QRPs, preregistration should logically increase confidence in research findings [53]. This enhanced credibility is particularly valuable for replication studies of candidate genes, where the historical context of false positives necessitates stronger evidence for claims.
Implementation Guide: Preregistration Workflows
Preregistration Process Visualization
The preregistration process follows a systematic workflow from initial planning through implementation and reporting. The diagram below illustrates key decision points and actions at each stage:
Template Selection Guide
Choosing the appropriate preregistration template is critical for effectively documenting research plans. OSF currently offers 11 different preregistration templates tailored to various research methodologies [56]. The selection process should match the template to the study design:
Table 3: Template Selection Guide for Different Research Types
| Research Type | Recommended Template | Key Elements | Considerations |
|---|---|---|---|
| General Quantitative | OSF Preregistration | Hypotheses, design, sample plan, variables, analysis strategy | Most comprehensive option; good for first-time users |
| Direct Replication | Replication Recipe (Pre-Study) | How study matches original, criteria for success | Essential for ensuring methodological fidelity |
| Existing Data Analysis | Preregistration for Studies with Existing Data | Knowledge of dataset, bias mitigation steps | Must document prior data exposure and preventive measures |
| Systematic Review | Generalized Systematic Review | Search strategy, inclusion/exclusion criteria, validation | Focuses on reproducible literature search methods |
| Qualitative Research | Qualitative Preregistration | Assumptions, beliefs, credibility strategies | Accommodates evolving data collection and analysis |
| Registered Reports | Registered Report Protocol | Manuscript with in-principle acceptance | For journals offering Registered Report format |
For researchers uncertain about template selection, practical strategies include downloading multiple templates to compare their questions before committing, using more concise templates like AsPredicted for exploratory research, or selecting the general OSF Preregistration template when no discipline-specific template aligns perfectly with the study [56].
Special Considerations for Replication Studies
Preregistering replication studies requires additional specific considerations beyond standard research documentation. For direct replications of candidate genes research, the Replication Recipe (Pre-Study) template provides a structured approach to articulate how the new study will match the original methodology and pre-specify criteria for determining whether results support the original findings [56]. This explicit specification is crucial for preventing flexible interpretation of replication success.
When working with existing data—common in genetic research utilizing biobanks—the Preregistration for Studies with Existing Data template helps researchers document their prior knowledge of the dataset and specify steps taken to mitigate analytical bias [56]. The template categorizes preregistration timing relative to data observation: before data collection, before human observation, before data access, or before analysis of specific variables [55]. This granular approach maintains the confirmatory value of analyses even when using pre-existing datasets.
Research Reagent Solutions: Essential Tools for Preregistration
Implementing effective preregistration requires leveraging available tools and resources. The following table catalogues key solutions that support various aspects of the preregistration process:
Table 4: Research Reagent Solutions for Preregistration and Transparency
| Tool/Resource | Primary Function | Application in Research | Key Features |
|---|---|---|---|
| OSF Registries | Public repository for preregistrations | Storing time-stamped research plans | Multiple templates, embargo options, versioning |
| AsPredicted | Streamlined preregistration | Simple confirmatory studies | Minimalist approach, essential questions only |
| Zenodo | General-purpose repository | Archiving preregistration documents | DOI assignment, long-term preservation |
| Registered Reports | Publication format | Results-blind manuscript review | In-principle acceptance, peer review before data collection |
| Transparent Changes Document | Deviation reporting | Documenting protocol modifications | Tracks and justifies changes from original plan |
| UKRN Preregistration Primer | Educational resource | Learning preregistration fundamentals | Discipline-specific guidance and examples |
Preregistration represents a fundamental shift in research practice that directly addresses the methodological limitations exposed by the replication crisis. For candidate genes research and replication studies, it offers a systematic approach to enhance transparency, reduce bias, and improve the credibility of scientific claims. The available evidence indicates that preregistration improves effect size estimation, facilitates more accurate power calculations for replication studies, and increases trust in research findings [61] [53].
While platforms vary in their features and suitability for different research contexts, OSF emerges as the most comprehensive solution for most researchers, offering template variety, workflow integration, and flexibility to accommodate different methodological approaches [56] [58]. The implementation of preregistration requires careful planning and template selection, but the benefits—including improved study design, protection against cognitive biases, and enhanced research credibility—substantially outweigh the initial time investment [60].
As the scientific community continues to prioritize research transparency and reproducibility, preregistration is poised to become standard practice across disciplines. For researchers investigating candidate genes, adopting preregistration represents not merely a procedural change, but a fundamental commitment to producing more reliable, trustworthy, and replicable scientific evidence.
In the field of genetic research on Premature Ovarian Insufficiency (POI), the replication of findings and validation of candidate genes are fundamental to establishing robust disease mechanisms and identifying viable therapeutic targets. The exponential growth in collaborative research, which trebled between 1990 and 2005, underscores the importance of standardized methodologies to ensure findings are reliable and reproducible [62]. Methodological fidelity—the degree to which an intervention or programme is delivered as intended by its developers—acts as a critical moderator between experimental interventions and their intended outcomes [63]. In POI research, where genetic heterogeneity is high and pathogenic variants in known genes account for only 18.7% to 23.5% of cases, the failure to implement studies with high fidelity can lead to Type III errors, where a lack of impact is erroneously attributed to the intervention itself rather than to poor implementation [63] [4]. This guide provides a structured framework for collaborating with original authors to ensure methodological fidelity in replication studies, objectively comparing approaches for validating POI candidate genes.
A Conceptual Framework for Implementation Fidelity in Research
Implementation fidelity is defined as the degree to which programs are implemented as intended by the program developers, a concept also termed "integrity" [63]. The conceptual framework for implementation fidelity comprises several core elements which are particularly relevant to experimental replication. Figure 1 illustrates the relationships between these elements and their collective role in achieving faithful implementation.
Figure 1: Framework for Implementation Fidelity. This diagram illustrates the core components of implementation fidelity and their interrelationships in experimental replication.
The framework positions Adherence as the central measure of implementation fidelity, encompassing several sub-elements [63]:
- Content: Whether all critical components of the experimental protocol are delivered.
- Frequency/Duration: Whether the intervention is administered at the prescribed intensity and length.
- Coverage: Whether all intended participants or samples receive the intervention as designed.
This adherence is moderated by three key factors [63]:
- Quality of Delivery: The skill and manner in which the experimental procedures are performed.
- Participant Responsiveness: The engagement and response of research subjects or biological materials to the intervention.
- Intervention Complexity: How complicated the original methodology is to implement, which can present substantial barriers to faithful replication.
Strategies to enhance fidelity include comprehensive documentation, training, and monitoring systems, which are categorized as Facilitation Strategies [63].
Comparative Analysis of Validation Approaches for POI Candidate Genes
Different methodological approaches offer distinct advantages and limitations for validating POI candidate genes. The choice of methodology depends on research goals, available resources, and the specific genetic variants under investigation. Table 1 provides a comparative analysis of primary validation approaches used in POI research.
Table 1: Comparison of Methodological Approaches for POI Candidate Gene Validation
| Method | Protocol Summary | Key Fidelity Parameters | Advantages | Limitations |
|---|---|---|---|---|
| Whole Exome Sequencing (WES) | High-throughput sequencing of protein-coding regions; variant calling against reference genomes [4]. | Read depth (>50x), coverage uniformity (>90%), variant calling accuracy [4]. | Unbiased approach; detects novel variants; comprehensive [4]. | Misses non-coding regions; requires robust bioinformatics. |
| Functional Validation (In Vitro) | Introduce candidate variants into cell lines; assess protein function, localization, or interaction [4]. | Expression vector identity, transfection efficiency, assay reproducibility. | Establishes causal mechanisms; controls confounding factors. | May not reflect in vivo context; overexpression artifacts. |
| Functional Validation (In Vivo) | Generate genetically modified mouse models; characterize ovarian phenotype and folliculogenesis [6]. | Genetic background consistency, phenotypic assessment standardization, environmental controls. | Models complex biology; assesses organism-level effects. | Time-consuming; expensive; species-specific differences. |
| Case-Control Association | Compare variant frequencies between well-characterized POI cases and matched controls [4]. | Case/control matching criteria, sequencing methodology consistency, statistical power. | Provides statistical evidence for association; uses human data. | Requires large sample sizes; establishes association not mechanism. |
Recent research demonstrates the power of these approaches. A 2023 study performing WES on 1,030 POI patients identified 195 pathogenic/likely pathogenic variants in 59 known POI-causative genes, accounting for 193 (18.7%) cases [4]. Association analyses further identified 20 novel POI-associated genes with a significantly higher burden of loss-of-function variants [4]. The genetic contribution was higher in primary amenorrhea (25.8%) than secondary amenorrhea (17.8%), highlighting how patient characteristics can influence research outcomes—a critical consideration for methodological fidelity [4].
Experimental Protocols for Key POI Validation Methods
Whole Exome Sequencing and Analysis Protocol
The following protocol is adapted from large-scale POI genetic studies [4]:
- DNA Extraction: Isolate genomic DNA from peripheral blood using standardized extraction kits; quantify using fluorometry and assess quality via spectrophotometry (A260/280 ratio ~1.8-2.0).
- Library Preparation & Exome Capture: Fragment DNA (150-200bp); perform end-repair, A-tailing, and adapter ligation. Hybridize to biotinylated oligonucleotide probes targeting exonic regions (e.g., Illumina Nexome, Agilent SureSelect).
- Sequencing: Perform high-throughput sequencing on Illumina platform (minimum 50x mean coverage, >90% of target bases covered at ≥20x).
- Variant Calling: Align reads to reference genome (GRCh38) using BWA-MEM; mark duplicates; call variants with GATK HaplotypeCaller.
- Variant Filtering & Annotation: Filter against population databases (gnomAD MAF <0.01); annotate functional impact with ANNOVAR/SnpEff.
- Pathogenicity Assessment: Classify variants according to ACMG guidelines; prioritize loss-of-function and predicted damaging missense variants.
Functional Validation via Homologous recombination Repair Assay
For genes involved in meiotic processes like HFM1, MCM8, MCM9, and MSH4, homologous recombination (HR) repair capacity can be functionally assessed [4]:
- Cell Culture & Transfection: Maintain HEK293T or other appropriate cell lines; transfect with plasmids carrying wild-type or candidate variant alleles.
- DNA Damage Induction: 48 hours post-transfection, induce DNA double-strand breaks with ionizing radiation (4-8 Gy) or chemical agents (e.g., 1μM camptothecin).
- HR Efficiency Measurement: Transfect with HR reporter (e.g., DR-GFP plasmid); measure GFP-positive cells via flow cytometry 72 hours post-transfection.
- Immunofluorescence Staining: Fix cells; stain for RAD51 foci (primary antibody anti-RAD51, secondary Alexa Fluor-conjugated); counterstain with DAPI; quantify foci/nucleus.
- Statistical Analysis: Compare HR efficiency and RAD51 foci formation between wild-type and variant groups using t-tests (≥3 independent experiments).
Visualization of Experimental Workflows
To ensure methodological fidelity in replication studies, clearly defined experimental workflows are essential. Figure 2 outlines the core pathway for validating novel POI candidate genes, from initial identification through functional confirmation.
Figure 2: POI Candidate Gene Validation Workflow. This diagram outlines the sequential process for identifying and validating POI candidate genes.
Following initial gene identification, specific experimental pathways are employed to establish biological mechanism. Figure 3 details the workflow for functionally characterizing genes involved in meiotic processes using homologous recombination repair assays.
Figure 3: Functional Validation Workflow for Meiotic Genes. This diagram details the experimental process for characterizing genes involved in homologous recombination repair.
The Scientist's Toolkit: Essential Research Reagents for POI Gene Validation
Faithful replication requires access to and proper implementation of critical research reagents. Table 2 catalogues essential materials and their functions for POI gene validation studies.
Table 2: Essential Research Reagents for POI Candidate Gene Validation
| Reagent/Resource | Function | Example Products/Sources | Critical Quality Controls |
|---|---|---|---|
| High-Quality DNA Samples | Source of genetic material for sequencing; fidelity depends on integrity and purity. | PAXgene Blood DNA Kit, QIAamp DNA Blood Maxi Kit | Concentration >50ng/μL; A260/280: 1.8-2.0; minimal degradation. |
| Whole Exome Capture Kits | Target enrichment for efficient sequencing of coding regions. | Illumina Nexome, Agilent SureSelect, IDT xGen | Coverage uniformity; on-target rate >70%; minimal capture bias. |
| POI-Specific Gene Panels | Focused screening of established POI genes; efficient for clinical validation. | Custom panels including NR5A1, MCM9, HFM1, EIF2B2, etc. | Content updated with new gene discoveries; coverage of all exons. |
| Expression Vectors | Delivery system for functional studies of candidate variants. | pcDNA3.1, pCMV, lentiviral vectors | Sequence verification; expression level quantification; empty vector controls. |
| HR Repair Reporters | Measure homologous recombination efficiency in functional assays. | DR-GFP, pCBAce3 plasmid systems | Proper calibration; positive and negative controls in each experiment. |
| Meiotic Markers | Visualize and quantify key meiotic processes in cellular models. | Anti-RAD51, Anti-γH2AX, Anti-SYCP3 antibodies | Antibody specificity validation; appropriate blocking controls. |
| Reference Controls | Ensure technical consistency across experiments and batches. | Coriell Institute samples; commercial controls | Include in every sequencing run/experimental batch; monitor for drift. |
The critical importance of these reagents is exemplified by the finding that genes implicated in meiosis or homologous recombination repair accounted for the largest proportion (48.7%) of detected cases in a recent POI cohort, highlighting the essential nature of validated reagents for studying these biological processes [4].
Ensuring methodological fidelity through collaborative practices with original authors is not merely a procedural concern but a fundamental scientific requirement in POI genetic research. The frameworks, comparisons, and standardized protocols presented here provide researchers with actionable strategies to enhance the rigor and reproducibility of their validation studies. As the genetic landscape of POI continues to expand—with recent discoveries adding 20 novel candidate genes to the existing 59 known causative genes—the commitment to implementation fidelity becomes increasingly critical for translating genetic findings into clinically relevant insights and therapeutic interventions [4].
Navigating Common Pitfalls and Limitations in Replication Efforts
Addressing Inadequate Statistical Power and Underestimated Sample Sizes
In the field of genetic research, particularly in the study of complex traits and disorders, the reliability of findings hinges on appropriate statistical power and adequate sample sizes. Inadequate power represents one of the most prevalent statistical errors in scientific studies, often leading to false negative results, overstatement of findings, and ultimately, reduced reproducibility [44]. Within the specific context of Premature Ovarian Insufficiency (POI) candidate gene research, these statistical challenges become particularly pronounced due to the condition's genetic heterogeneity and the typically small sample sizes available for study.
Statistical power, defined as the probability of correctly rejecting a false null hypothesis (typically calculated as 1-β), is critically influenced by sample size, effect size, and alpha level [44]. The ideal power for a study is generally considered to be 0.8 (or 80%), yet many genetic studies, especially initial candidate gene investigations, fall substantially short of this benchmark [44]. This inadequate power has direct implications for POI research, where identifying genuine genetic associations amidst multiple testing burdens requires careful statistical planning.
The consequences of underpowered studies extend beyond scientific validity to encompass ethical considerations, including time loss, cost inefficiencies, and the potential misuse of limited research resources [44]. As we examine the current landscape of POI genetic research, it becomes evident that addressing these statistical shortcomings is not merely a methodological concern but a fundamental requirement for advancing our understanding of this complex condition.
The Current Landscape: Sample Sizes in Recent Genetic Studies
Recent advances in genetic research methodologies have enabled increasingly large-scale studies, yet significant variation exists in sample sizes across different research contexts. The table below summarizes the sample sizes employed in several recent genetic studies, providing context for typical cohort sizes in contemporary genetic research:
Table 1: Sample Sizes in Recent Genetic Studies
| Study Focus | Sample Size | Study Type | Key Findings | Reference |
|---|---|---|---|---|
| Premature Ovarian Insufficiency (POI) | 1,030 patients; 5,000 controls | Whole-exome sequencing | Identified pathogenic variants in 59 known POI-causative genes in 18.7% of cases | [4] |
| Delay Discounting (Impulsivity) | 134,935 participants | Genome-wide association study (GWAS) | Identified 11 genetic regions linked to delay discounting | [64] |
| Brief Resilience Scale | 124,774 participants | Genome-wide association study (GWAS) | First GWAS of BRS, SNP-based heritability of 7.3% | [65] |
| Primary Ovarian Insufficiency | 291 cases; 233 controls | Whole-exome sequencing | Identified candidate causative variants through WES | [66] |
The disparity in sample sizes between large-scale biobank studies and disease-specific cohorts like POI research is striking. While GWAS of behavioral traits can leverage samples exceeding 100,000 participants [65] [64], the largest POI whole-exome sequencing study to date included 1,030 patients [4]. This sample size discrepancy directly impacts the ability to detect genetic associations, particularly for rare variants with moderate effect sizes.
The POI study with 1,030 cases represented a substantial advancement in the field, enabling the identification of 195 pathogenic/likely pathogenic variants across 59 known genes [4]. Nevertheless, even this comparatively large cohort left approximately 76.5% of cases without definitive genetic explanations, highlighting the need for even larger samples to uncover the full spectrum of genetic contributors to POI [4]. The relationship between sample size and genetic discovery is further complicated by the heterogeneity of POI, which encompasses a clinical continuum from primary amenorrhea to early menopause [66].
Statistical Foundations: Calculating Appropriate Sample Sizes
Key Statistical Concepts and Relationships
The relationship between sample size, statistical power, effect size, and significance level forms the foundation of appropriate study design. Understanding these interconnections is essential for avoiding both underpowered studies and ethically problematic redundant sampling [44].
The most common alpha level (α) chosen in scientific studies is 0.05, representing a 5% risk of concluding that an effect exists when it actually does not (Type I error) [44]. However, this standard level may be inappropriate for certain research contexts. In pilot studies, α is often set at 0.10 or 0.20, while in studies where incorrectly concluding effectiveness could have dangerous consequences (such as drug studies), alpha may be set much lower at 0.001 or less [44].
The P value represents the obtained statistical probability of incorrectly accepting the alternate hypothesis and is compared against the predetermined alpha level to determine statistical significance [44]. Equally important is the Type II error (β), which represents the probability of incorrectly rejecting H1 and wrongly accepting H0 (false negative) [44]. Power is calculated as 1-β, with the ideal power for a study considered to be 0.8 (80%) [44].
Table 2: Key Statistical Concepts in Sample Size Determination
| Statistical Concept | Definition | Impact on Sample Size | Common Standards |
|---|---|---|---|
| Alpha (α) | Probability of Type I error (false positive) | Lower α requires larger sample size | 0.05 (general research), 0.001 (high-risk studies) |
| Beta (β) | Probability of Type II error (false negative) | Lower β requires larger sample size | 0.20 (for 80% power) |
| Power (1-β) | Probability of correctly detecting a true effect | Higher power requires larger sample size | 0.80 (ideal minimum) |
| Effect Size (ES) | Magnitude of the phenomenon being studied | Smaller effect sizes require larger sample sizes | Varies by research field |
| P Value | Obtained probability of incorrectly accepting H1 | Compared to α to determine significance | < 0.05 considered statistically significant |
Practical Sample Size Calculation Methods
Different research questions require specific methodological approaches to sample size calculation. The formulas vary based on study design, with each addressing distinct statistical requirements:
For survey-type studies estimating proportions, the sample size can be calculated using: N = (Zα/2^2 * P(1-P) * D) / E^2 Where N is sample size, P is prevalence or proportion of event, E is precision (margin of error), D is design effect, and Zα/2 is 1.96 for alpha 0.05 [44].
For studies comparing two means: n1 = (r + 1)/r * (σ/d)^2 * (Z1-β + Zα/2)^2 Where r = n1/n2 (the ratio of sample size), σ is pooled standard deviation, d is difference of means of two groups, Z1-β is 0.84 for power 0.80, and Zα/2 is 1.96 for alpha 0.05 [44].
For studies comparing two proportions: n = (Zα/2 + Z1-β)^2 * (p1(1-p1) + p2(1-p2)) / (p1 - p2)^2 Where p1 and p2 are proportions of events for group I and group II [44].
These calculations can be performed manually, using nomograms, or through specialized software [44]. The Figure 2 nomogram referenced in the search results represents one commonly used tool for sample size estimation using effect size and power [44].
Diagram 1: Relationships among statistical power concepts. This diagram illustrates how sample size, effect size, and alpha level interact to influence statistical power and error rates in research design.
Case Study: Power Considerations in POI Candidate Gene Research
Genetic Architecture and Statistical Challenges
Premature Ovarian Insufficiency presents unique challenges for genetic analysis due to its heterogeneous etiology and the diverse inheritance patterns observed in associated genes. Twin studies estimate the heritability of POI at 53% to 71%, and there is a strong relationship between age at menopause in mothers and daughters, with an odds ratio of 6 for early menopause in daughters whose mothers had early menopause [66]. Despite this strong genetic component, known genetic causes explain less than 30% of POI cases [66].
The genetic architecture of POI encompasses various modes of inheritance, including autosomal recessive, autosomal dominant, and X-linked patterns [4]. In the large POI study of 1,030 patients, most cases (80.3%) with identified pathogenic variants carried monoallelic (single heterozygous) variants, while 12.4% had biallelic variants, and 7.3% had multiple pathogenic variants in different genes [4]. This heterogeneity directly impacts power calculations, as different variant frequencies and effect sizes require tailored statistical approaches.
The sample size requirement varies significantly between different genetic analyses. For establishing single-gene associations through case-control burden testing, large cohorts are essential to achieve statistical significance after multiple testing corrections. In the discovery of 20 novel POI-associated genes, the study required 1,030 cases and 5,000 controls to detect a significantly higher burden of loss-of-function variants [4]. Even with this substantial sample size, the identified genes collectively explained only an additional portion of cases beyond the 18.7% explained by known genes [4].
Sample Size Implications for Validation Studies
In replication studies of POI candidate genes, power considerations become particularly important. The validation of candidate genes requires careful attention to sample size calculations based on the expected effect sizes and allele frequencies observed in initial discovery studies.
For rare variant associations, such as the heterozygous mutations identified in known POI genes like NR5A1 and MCM9 (each found in approximately 1.1% of patients) [4], replication cohorts must be sufficiently large to detect similarly rare effects. The statistical power to validate such findings depends not only on the sample size but also on the genetic architecture of the specific candidate gene.
The difference in genetic contribution between clinical subtypes also has implications for power calculations in validation studies. Patients with primary amenorrhea showed a higher overall contribution of pathogenic variants (25.8%) compared to those with secondary amenorrhea (17.8%) [4]. Furthermore, a considerably higher frequency of biallelic and multi-heterozygous pathogenic variants was observed in patients with primary amenorrhea [4]. This suggests that replication studies focusing on specific POI subtypes may require different sample sizes based on the expected genetic architecture.
Practical Implementation: Protocols for Power Calculation in Genetic Studies
Step-by-Step Protocol for Sample Size Determination
Implementing appropriate power calculations requires a systematic approach to ensure all relevant factors are considered. The following protocol provides a framework for determining sample sizes in genetic studies:
Define Primary Hypothesis and Outcome Measures: Clearly specify the genetic model (dominant, recessive, additive) and the primary outcome (variant burden, allele frequency, etc.). In POI research, this typically involves defining the specific genetic association to be tested [4].
Establish Significance Threshold: Determine the alpha level appropriate for your study context. For genetic studies with multiple testing burdens, consider implementing stricter alpha levels or correction methods [44].
Estimate Expected Effect Size: Base effect size estimates on prior studies or pilot data. For novel POI gene discovery, consider effects observed in similar genes. The large POI study identified genes with varying prevalence, from high-frequency contributors like EIF2B2 (0.8%) to rarer associations [4].
Select Appropriate Power Level: Choose the desired statistical power, with the ideal being 80% or higher [44]. For preliminary studies, lower power might be acceptable, while for definitive validation studies, higher power is preferable.
Calculate Required Sample Size: Apply the appropriate formula or software based on your study design. For genetic association studies, specialized power calculation tools that account for genetic parameters (allele frequency, linkage disequilibrium, etc.) are recommended [44].
Account for Practical Constraints: Consider recruitment feasibility, budget limitations, and ethical considerations. In POI research, the rarity of the condition may limit achievable sample sizes, necessitating collaborative consortia [44] [4].
Document Justification: Thoroughly document all assumptions, parameters, and calculations used in sample size determination to ensure transparency and reproducibility.
Research Reagent Solutions for Genetic Studies
Table 3: Essential Research Reagents and Materials for POI Genetic Studies
| Reagent/Material | Function/Application | Example Use in POI Research |
|---|---|---|
| Whole Exome Sequencing Kits (e.g., NimbleGen, SureSelect) | Target enrichment for exome sequencing | Capturing exonic regions for variant identification in POI cohorts [4] |
| Whole Genome Sequencing Platforms (e.g., Illumina X Ten) | Comprehensive variant discovery across entire genome | Control sample sequencing in POI studies [66] |
| Variant Caller Software (e.g., Sentieon, BWA-MEM) | Alignment and variant calling from sequencing data | Processing WES data from POI patients and controls [66] [4] |
| Variant Annotation Tools (e.g., VAAST, VVP) | Prioritizing potentially deleterious variants and damaged genes | Scoring VCF files to identify pathogenic variants in POI cases [66] |
| Functional Validation Reagents (D. melanogaster models) | Experimental validation of candidate gene pathogenicity | Functional studies on identified genes not previously implicated in POI [66] |
Diagram 2: Genetic research workflow for POI studies. This diagram outlines the key stages in genetic association studies, from initial design through validation, with statistical power considerations impacting each phase.
The challenge of inadequate statistical power and underestimated sample sizes remains a significant concern in POI candidate gene research. As the field advances, several key strategies emerge for addressing these limitations.
First, collaborative consortia and data sharing are essential for accumulating sample sizes sufficient to detect genetic effects with moderate to small effect sizes. The success of large-scale biobank studies in identifying genetic associations for complex traits demonstrates the power of such approaches [65] [64]. For rare conditions like POI, international collaborations are particularly valuable for achieving statistically meaningful cohort sizes.
Second, careful attention to power calculations during study design phase is crucial. Researchers should base sample size determinations on realistic effect size estimates, appropriate significance thresholds, and desired power levels [44]. Transparent reporting of power calculations and their assumptions enhances the interpretability and reproducibility of research findings.
Finally, the integration of functional validation approaches helps mitigate the risk of false positive associations, particularly in studies with limited statistical power [66] [4]. The combination of statistical evidence with biological validation strengthens the evidence for candidate genes identified in underpowered studies.
As POI research continues to evolve, addressing these statistical challenges will be essential for advancing our understanding of this complex condition and developing improved approaches for diagnosis, counseling, and potential therapeutic interventions.
The reproducibility of experimental findings is a cornerstone of the scientific method, serving as the ultimate test for the validity of empirical claims. However, across numerous scientific disciplines—from psychology to genetics—researchers frequently encounter discrepant results when attempting to replicate previously established findings. The interpretation of these discrepancies often centers on a critical distinction: do they represent a true failure to replicate (indicating the original finding may have been a false positive), or do they reflect the influence of hidden moderators (contextual factors that meaningfully alter the effect)? Within genetics research, particularly in the study of candidate genes for complex traits and disorders, this distinction carries profound implications for the allocation of research resources, the development of pharmacological interventions, and the fundamental understanding of disease etiology.
The replication crisis in psychology has brought this issue into sharp focus. The Reproducibility Project: Psychology, a large-scale effort to replicate 100 studies published in top psychology journals, found that only 39% could be unambiguously reproduced [67]. This landmark project ignited intense debate about how to interpret such widespread failures to replicate. Similar challenges are now emerging in genetic association studies, where initial exciting findings often fail to replicate in subsequent, larger studies, prompting questions about what distinguishes robust genetic associations from spurious ones.
This guide provides a structured framework for comparing these two competing interpretations, offering methodological tools to distinguish between true replication failures and effects moderated by unseen variables, with specific application to candidate gene research in drug development.
Theoretical Framework: Defining the Competing Interpretations
True Failure vs. Hidden Moderators: Conceptual Distinctions
The interpretation of discrepant replication results typically falls into one of two competing categories, each with distinct implications for research validity and progression.
Table 1: Conceptual Comparison of True Failure vs. Hidden Moderators
| Aspect | True Failure Interpretation | Hidden Moderators Interpretation |
|---|---|---|
| Core Definition | Original finding was a false positive resulting from methodological or statistical artifacts | Original finding is context-dependent and varies due to unmeasured variables |
| Primary Cause | Questionable research practices, low statistical power, publication bias, analytical flexibility | Genuine biological or environmental heterogeneity that systematically alters effects |
| Implication for Original Finding | Invalid | Conditionally valid |
| Research Response | Abandon hypothesis or improve methodological rigor | Identify boundary conditions and discover moderating variables |
| Example in Genetics | Initial candidate gene association fails due to inadequate multiple testing correction | Genetic effect varies across populations due to ethnicity-specific genetic backgrounds |
The true failure interpretation suggests that the original finding was never genuine, but rather emerged from a combination of methodological weaknesses. In genetics research, this might include inadequate sample sizes, population stratification, incomplete correction for multiple testing, or analytical flexibility (e.g., p-hacking) [68]. From this perspective, failed replications correctly identify the absence of a real effect.
In contrast, the hidden moderators interpretation proposes that the original finding reflects a genuine but context-dependent relationship. Here, the failure to replicate stems from differences between the original and replication contexts that meaningfully alter the effect. In genetics, such moderators might include population-specific genetic backgrounds, gene-environment interactions, epigenetic factors, or developmental stages [67]. The Reproducibility Project: Psychology found empirical support for this perspective, demonstrating that the contextual sensitivity of research topics was significantly associated with replication success, even after adjusting for methodological characteristics [67].
Historical Context and Evolution of the Debate
The tension between these interpretations is not new. In 1981, social psychologist John Touhey criticized a failed replication based on the "dubious ... assumption that experimental manipulations can be studied apart from the cultural and historical contexts that define their meanings" [67]. This perspective finds its philosophical roots in Kurt Lewin's equation that behavior is a function of both the person and the environment (B = f(P,E)) [67].
The debate has intensified with psychology's replication crisis, but similar challenges exist across scientific domains. As one researcher notes, "Many rodent studies are doomed to irreproducibility because subtle environmental differences, such as food, bedding, and light, can affect biological and chemical processes that determine whether experimental treatments succeed or fail" [67]. Even Isaac Newton contended that failed replications of his color spectrum research resulted from his contemporaries using inferior prisms [67].
Methodological Approaches for Distinguishing Interpretations
Experimental Designs for Testing Competing Hypotheses
Different research designs provide varying capabilities to distinguish between true failures and hidden moderators. The table below compares key methodological approaches.
Table 2: Methodological Approaches for Investigating Replication Discrepancies
| Methodology | Key Features | Strength for Distinguishing Interpretations | Practical Limitations |
|---|---|---|---|
| Direct Replication | Exact repetition of original methods | Establishes baseline reproducibility | Cannot identify specific moderators if failure occurs |
| Adversarial Collaboration | Opposing research teams jointly design studies | Directly tests competing interpretations through shared methodology | Requires substantial cooperation between teams |
| Multisite Replication | Simultaneous replications across multiple labs | Distinguishes random variation from systematic moderators | Resource-intensive |
| Constructive Replication | Different methods testing same hypothesis | Tests theoretical robustness beyond specific methodology | Cannot pinpoint why original method fails |
| Registered Reports | Peer review before data collection | Eliminates publication bias and questionable practices | Requires early commitment to publication |
The adversarial collaboration approach deserves special attention, as it provides a powerful framework for resolving replication disputes. In this model, researchers with different theoretical perspectives or conflicting findings jointly design studies to test competing interpretations. A notable example comes from research on the minimal group paradigm, where researchers with discrepant findings collaborated to systematically test potential hidden moderators [69]. Their partnership began with detailed comparison of their respective methodologies, identifying subtle differences that might function as moderators, then jointly designing studies to test these candidates empirically.
Statistical Framework for Interpretation
Statistical approaches play a crucial role in distinguishing true failures from hidden moderators. Key considerations include:
Power and sample planning: Many failed replications, particularly in early candidate gene studies, suffered from inadequate statistical power. Underpowered studies cannot reliably distinguish between true null effects and substantially smaller (but potentially meaningful) effects.
Heterogeneity testing: Significant between-study heterogeneity in meta-analyses often signals potential moderators rather than uniform false positives.
Bayesian approaches: These methods allow formal comparison of the relative evidence for null versus moderated effects, moving beyond simplistic dichotomous significance testing.
The Reproducibility Project found that after statistically adjusting for methodological characteristics like effect size and statistical power, contextual sensitivity remained a significant predictor of replication success [67]. This suggests that contextual factors explain variance in replication outcomes beyond what can be attributed to methodological quality alone.
Case Studies Across Disciplines
Psychology: The Minimal Group Paradigm
The minimal group paradigm (MGP)—which examines ingroup favoritism based on arbitrary group distinctions—provides an instructive case study of how adversarial collaboration can resolve replication discrepancies. Researchers who had obtained conflicting results with the MGP initiated a collaboration to identify potential hidden moderators [69].
Their process began with systematically comparing their methodological approaches, identifying numerous subtle differences in procedure. These included whether participants were socially isolated during the experiment, whether instructions were delivered live or via text, and the specific allocation measures used. Through joint experimentation, they empirically tested which of these methodological differences functioned as meaningful moderators of the effect.
This collaborative approach transformed what might have remained a contentious debate about failed replications into a productive investigation of boundary conditions and moderating factors. The researchers noted that such collaborations require researchers to compare protocols "in close and very explicit detail, with an emphasis on identifying taken-for-granted procedures and methodological differences that might be psychologically important" [69].
Genetics: Candidate Gene Studies
The field of genetics provides compelling examples of both true failures and hidden moderators in replication attempts. The early candidate gene era was marked by numerous promising associations that subsequently failed to replicate in larger, better-designed studies. Many of these represent true failures stemming from inadequate statistical correction for multiple testing and publication bias favoring positive results.
However, more recent research has identified genuine hidden moderators in genetic associations. For instance, a genome-wide association study of delay discounting (the tendency to prefer smaller immediate rewards over larger delayed ones) identified 11 genetic regions and found that the same genetic factors influencing impulsive decision-making also overlap with risks for metabolic conditions [64]. This suggests that some earlier failures to replicate genetic associations for impulsivity might have reflected unaccounted pleiotropic effects or developmental moderators.
Similarly, a multi-trait GWAS of lymphoid neoplasms employed sophisticated clustering approaches to identify shared genetic susceptibility across cancer subtypes, demonstrating how apparently inconsistent associations across studies might reflect disease heterogeneity rather than true failures [70].
Diagram 1: Framework for interpreting replication discrepancies, showing the two primary interpretations and their common causes.
Decision Framework for Candidate Gene Research
Evaluating Evidence for Competing Interpretations
Researchers facing discrepant results in candidate gene studies can apply the following systematic framework to evaluate the relative support for each interpretation:
Table 3: Decision Framework for Interpreting Genetic Replication Failures
| Evidence Type | Supports True Failure | Supports Hidden Moderators |
|---|---|---|
| Methodological Quality | Original study has low power, multiple testing issues | Both original and replication are methodologically rigorous |
| Consistency Pattern | Failures are consistent across diverse replication attempts | Failures show systematic variation (e.g., by population) |
| Meta-analytic Evidence | Effect size estimates converge toward null in cumulative meta-analysis | Significant between-study heterogeneity persists |
| Biological Plausibility | No coherent mechanism connects gene to phenotype | Plausible mechanisms exist for context-dependence |
| A Priori Predictions | Moderator explanations emerge only after replication failure | Boundary conditions were predicted theoretically |
Adversarial Collaboration Protocol for Genetic Research
Building on successful examples from psychology, we propose a structured protocol for implementing adversarial collaboration in genetic research:
Diagram 2: Adversarial collaboration workflow for resolving genetic replication disputes.
Research Reagent Solutions for Replication Research
Advanced methodological tools and resources are essential for conducting rigorous replication studies and investigating potential moderators.
Table 4: Essential Research Reagents for Genetic Replication Studies
| Reagent/Tool | Primary Function | Application in Replication Research |
|---|---|---|
| UK Biobank Data | Large-scale genetic and phenotypic data | Provides well-powered replication cohorts with diverse measures |
| PheWAS Catalog | Database of genotype-phenotype associations | Facilitates assessment of pleiotropy and cross-trait associations |
| METAL Software | Meta-analysis of genome-wide association studies | Enables coordinated analysis across multiple cohorts |
| LD Score Regression | Estimates heritability and genetic correlations | Quantifies shared genetic architecture between traits |
| GWAS Catalog | Curated collection of all published GWAS | Provides context for interpreting novel associations |
| FINEMAP Software | Bayesian fine-mapping of causal variants | Distinguishes causal variants from linked markers |
| Polygenic Risk Scores | Aggregate genetic susceptibility across many variants | Tests genetic effects across different populations |
These tools enable more nuanced approaches to replication that move beyond simple binary success/failure determinations. For example, a genome-wide association study of resilience using the Brief Resilience Scale in the UK Biobank demonstrated strong genetic correlations with neuroticism, depression, and anxiety, suggesting that apparent failures to replicate specific resilience associations might reflect measurement heterogeneity rather than absence of genetic effects [65].
Similarly, multi-trait GWAS approaches that leverage pleiotropy among biologically related phenotypes have improved discovery power in genetically heterogeneous traits like lymphoid neoplasms [70]. These methods explicitly account for the fact that genetic effects often manifest differently across related biological contexts.
The dichotomy between "true failure" and "hidden moderators" represents a false dichotomy when applied rigidly. Many replication discrepancies likely reflect a combination of both methodological limitations and genuine context-dependence. The most productive approach moving forward recognizes that:
Methodological rigor is non-negotiable for distinguishing these interpretations—well-powered, pre-registered studies with transparent analysis plans provide the essential foundation.
Theoretical sophistication should guide replication attempts, with explicit consideration of plausible moderators based on biological mechanisms.
Collaborative norms that encourage adversarial partnerships and multi-team replication efforts will accelerate resolution of conflicting findings.
Measurement invariance across studies must be established before comparing effect sizes—apparent replication failures may reflect methodological differences rather than true discrepancies.
As genetic research increasingly focuses on gene-environment interactions, developmental processes, and cross-population generalization, the recognition and systematic investigation of hidden moderators will become increasingly central to the field. By adopting the frameworks and methodologies outlined in this guide, researchers can transform contentious replication failures into opportunities for discovering the boundary conditions and contextual factors that give genetic associations their meaning and potential translational value.
The molecular processes underlying human health and disease are highly complex. Often, genetic and environmental factors contribute to a given disease or phenotype in a non-additive manner, yielding gene-environment (G × E) interactions [71]. Alongside interaction, gene-environment correlation (rGE) represents another crucial facet of gene-environment interplay, occurring when "genotypes are selectively exposed to different environments" [72]. Understanding these dynamics is particularly crucial in replication studies of candidate genes for conditions like Premature Ovarian Insufficiency (POI), where disentangling true genetic effects from environmental contingencies can determine success or failure in identifying valid therapeutic targets [73] [51]. The challenge lies in distinguishing between different types of rGE and G×E, each requiring specific methodological approaches for accurate detection and interpretation.
Theoretical Frameworks and Models
Types of Gene-Environment Correlation (rGE)
Typically, three types of rGE are distinguished, each with distinct mechanisms and implications for research design [72]:
Passive rGE: Results from parents providing their children with both genes and environments that are correlated with genetically-influenced parental characteristics. For example, parents with internalizing problems may both pass on genetic risk and create an anxious environment through emotional overinvolvement.
Evocative rGE: Occurs when a child's inherited characteristics evoke responses from their environment. For instance, children with internalizing problems may elicit emotional overinvolvement from their parents through their anxious behaviors.
Active rGE: Refers to children actively selecting environments for genetically influenced reasons. This type is less relevant for parent-child relationships as parents and children do not select one another.
Models of Gene-Environment Interaction (G×E)
Several theoretical models describe different patterns of G×E effects, each with distinct implications for research and interpretation [73]:
Diathesis-Stress Model: Suggests individuals with high genetic risk are more likely to experience adverse outcomes when exposed to high-risk environments. This represents the dominant interaction pattern (89.5%) in substance use studies with significant interaction effects [73].
Differential Susceptibility Model: Indicates that individuals with certain genetic predispositions have greater vulnerability to high-risk environments but can also obtain more benefits from positive environments [73].
Social Push Model: Implies genetic factors have strong effects at low or medium environmental risk levels, with social influences dominating in extreme environments [73].
Swing Model: Suggests individuals with medium genetic risk are most influenced by environmental factors in developing health risk behaviors [73].
Methodological Approaches and Experimental Designs
Traditional Quantitative Genetic Designs
Children-of-Twins (CoT) Models provide an effective strategy for disentangling direct environmental influences of parenting from passive rGE [72]. This design compares children of MZ twins, who share half their genes with both their parent and the parent's co-twin, with children of DZ twins, who share approximately 25% of their genes with their parent's co-twin. Since the rearing environment is distinct for each child, environmental effects can be estimated while controlling for genetic correlations between parents and children [72]. However, even this design has limited power for detecting evocative rGE due to modest variation in genetic relatedness among cousins [72].
Adoption Studies comparing associations between family environment and children's development in nonadoptive and adoptive families provide evidence for passive rGE, as these associations are typically stronger in biological families [74]. These designs help control for passive rGE by separating genetic relatedness from environmental exposure.
Genomic Era Methodological Advances
The synthesis of quantitative genetics and molecular genetics has transformed G×E research through quantitative genomics, which enables direct measurement of inherited DNA differences [74]. Key advancements include:
Polygenic Scores (PGS): Composites that aggregate thousands of tiny effects from genome-wide association studies, enabling genetic influence assessment in unrelated individuals without twins or adoptees [74]. Including parental polygenic scores in "trio" designs allows controlling for passive rGE when examining direct effects of children's polygenic scores [74].
Genome-Wide Interaction Studies (GWIS): Traditionally test G×E through linear regression (Y = β₀ + β₁G + β₂E + β₃G×E + ε), though this approach often suffers from low power due to collinearity between G and G×E [75].
Mendelian Randomization (MR) Framework: A powerful new approach conceptually connecting G×E with MR framework, where testing horizontal pleiotropy can be used for detecting G×E [75]. This method identifies genetic variants that depart from the expected regression line between marginal and main effects, indicating G×E or mediation.
Experimental Protocols for Candidate Gene Replication
For replication studies of POI candidate genes, comprehensive protocols should include:
Multi-Omics Integration: Combining RNA sequencing data from relevant tissues (e.g., granulosa cells for POI, endometrial tissue for RSA) with machine learning algorithms to identify hub genes [51]. This typically involves differential gene expression analysis, protein-protein interaction networks, transcription factor regulatory networks, and functional annotation.
Comprehensive Functional Validation: Including in vitro and in vivo models to test identified candidate genes. For POI and RSA research, this involves collecting granulosa cells from patients undergoing IVF/ICSI and endometrial tissues from patients with recurrent miscarriage, followed by qRT-PCR validation of hub genes [51].
Power Calculations and Covariate Control: Essential for reliable G×E detection, as underpowered studies and unmeasured confounding contribute to non-replication [73]. Studies should control for key covariates like steroid medication use, uterine organic lesions, autoimmune diseases, and history of radiotherapy/chemotherapy [51].
Quantitative Data in Gene-Environment Interplay
Empirical Evidence for rGE and G×E Across Domains
Table 1: Evidence for Gene-Environment Interplay Across Research Domains
| Research Domain | rGE Evidence | G×E Evidence | Key Findings | Methodological Approach |
|---|---|---|---|---|
| Parenting & Child Adjustment [72] | Strong | Moderate | Maternal emotional overinvolvement & child internalizing problems best explained by evocative rGE | Children-of-twins model |
| Early Life Cognitive Development [76] | Widespread | Minimal | Environmental measures accounted for 20.6% of variance vs. 0.5% for PGS; additive rather than interactive effects | Polygenic scores with 39 environmental measures |
| Substance Use Behavior [73] | Not reported | Strong (89.5% of studies) | Diathesis-stress model dominant (89.5%); differential susceptibility less common | Candidate gene-environment interaction |
| Serum Lipids [75] | Not reported | Confirmed | 5 loci (6 independent signals) interacting with smoking/alcohol consumption | Mendelian randomization framework |
Success Rates by Genetic Pathway in Substance Use Research
Table 2: Candidate Gene G×E Success Rates in Substance Use Research
| Genetic Pathway | Gene | Studies with Significant G×E | Dominant Interaction Model |
|---|---|---|---|
| Serotoninergic | 5-HTTLPR | 61.5% | Diathesis-stress |
| Monoaminergic | MAOA | 100% | Diathesis-stress |
| Dopaminergic | DRD2 | 42.9% | Diathesis-stress |
| Dopaminergic | DRD4 | 50% | Diathesis-stress |
| Dopaminergic | DAT | 50% | Diathesis-stress |
| Stress Response | CRHR1 | 80% | Diathesis-stress |
| Opioid System | OPRM1 | 100% | Diathesis-stress |
| GABAergic | GABRA1 | 100% | Diathesis-stress |
| Cholinergic | CHRNA | 50% | Diathesis-stress |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Materials for Gene-Environment Interplay Studies
| Research Reagent | Specific Examples | Function in G×E Research |
|---|---|---|
| Genotyping Platforms | DNA microarrays ("SNP chips") [74] | Genotyping hundreds of thousands of DNA variants for genome-wide association studies |
| Polygenic Score Calculators | PRSice, LDpred [74] | Aggregating thousands of tiny SNP effects into composites for genetic risk assessment |
| Transcriptomic Datasets | GEO database resources [51] | Providing RNA sequencing data from target tissues (e.g., granulosa cells, endometrial tissue) |
| Bioinformatic Tools | Cytoscape, machine learning algorithms [51] | Constructing protein-protein interaction networks and identifying hub genes |
| Functional Validation Assays | qRT-PCR protocols [51] | Validating identified hub genes in relevant cell and tissue samples |
| Environmental Assessment Tools | Home and neighborhood environment measures [76] | Quantifying environmental exposures for correlation and interaction analyses |
Analytical Framework for POI Candidate Gene Replication
For replication studies of POI candidate genes, specific analytical workflows are essential to account for gene-environment interplay:
This framework emphasizes that successful replication requires progressing through sequential analytical phases, each addressing specific challenges in gene-environment interplay. The multi-omics integration phase identifies potential candidate genes through analysis of granulosa cells from POI patients and endometrial tissue from RSA patients [51]. The rGE assessment phase determines whether genetic factors influence environmental exposures relevant to POI, such as autoimmune triggers or chemical exposures [74]. The G×E testing phase examines whether genetic effects on POI susceptibility are moderated by specific environmental factors [71]. Finally, functional validation confirms biological mechanisms in relevant cellular models [51].
Understanding gene-environment interplay remains both a challenge and opportunity in complex disease research. For POI candidate gene replication studies, successful navigation of rGE and G×E complexities requires sophisticated designs that go beyond simple association testing. The integration of polygenic scores with robust environmental measures, application of novel analytical frameworks like Mendelian randomization for G×E detection, and comprehensive functional validation represent promising paths forward [74] [75]. Future research should prioritize large-scale collaborative studies with diverse populations, precise environmental measurement, and multi-omics integration to fully elucidate how genetic predispositions and environmental factors jointly influence disease risk across the life course.
The long-standing paradigm in genetics, which focused primarily on single-gene (monogenic) disorders, has progressively expanded to encompass the complex spectrum of polygenic and omnigenic models. This evolution is particularly relevant in the study of Premature Ovarian Insufficiency (POI), where nearly 70% of cases remain etiologically unexplained despite extensive investigation of monogenic causes [45]. Traditional monogenic approaches have successfully identified pathogenic variants in genes such as NOBOX, BMP15, and GDF5 in a subset of POI cases, but this model fails to explain the majority of idiopathic cases [45].
The emerging understanding of complex traits suggests that disease-causing variants are spread across most of the genome rather than clustering exclusively into key disease pathways [77]. This "omnigenic" perspective proposes that gene regulatory networks are sufficiently interconnected that all genes expressed in disease-relevant cells can potentially affect the functions of core disease-related genes [77]. For conditions like POI, this model provides a more comprehensive framework for understanding the dispersed genetic architecture that likely underlies most cases. As we move beyond single-gene effects, researchers are increasingly adopting both polygenic risk scores and sophisticated general/specific models to capture the full genetic liability for complex conditions.
Comparative Analysis of Genetic Models
Defining the Spectrum of Genetic Inheritance
Table 1: Characteristics of Genetic Inheritance Models
| Model Type | Genetic Basis | Inheritance Patterns | Example Conditions | Research Approaches |
|---|---|---|---|---|
| Monogenic | Single gene variants | Mendelian (autosomal dominant/recessive, X-linked) | Huntington's Disease, Cystic Fibrosis [78] [79] | Candidate gene sequencing, Family studies |
| Oligogenic | Primary gene with modifier genes | Modified Mendelian | Spinal Muscular Atrophy (SMN1 with SMN2 modifiers) [79] | Targeted gene panels, Copy number variation analysis |
| Polygenic | Multiple genes with small effects | Complex, multifactorial | Coronary artery disease, Height, Schizophrenia [78] [77] | Genome-wide association studies (GWAS), Polygenic risk scores |
| Omnigenic | Core genes + peripheral genes across regulatory networks | Highly complex, diffuse | Idiopathic POI, Many common complex diseases [77] | Integration of GWAS with functional genomics, Network analyses |
Quantitative Comparison of Genetic Contributions
Table 2: Empirical Performance of Genetic Models in Complex Trait Analysis
| Model Feature | Single-Gene/ Oligogenic Models | Polygenic Models | Evidence Source |
|---|---|---|---|
| Variance Explained | High for specific cases (e.g., FIGLA in POI: 1 patient in 28) [45] | Common variants: ~13% (median across 22 traits) [80] | UK Biobank exome sequencing (394,783 individuals) [80] |
| Rare Variant Contribution | 28.6% of POI cases with causal SNV/indel [45] | Rare coding variants: 1.3% (average across 22 traits) [80] | Burden Heritability Regression analysis [80] |
| Diagnostic Yield in POI | Combined array-CGH and NGS: 57.1% (16/28 patients) [45] | Not yet routinely applied in clinical POI diagnosis | Idiopathic POI cohort study [45] |
| Model Flexibility | Fixed biological assumptions (additive, dominant, recessive) [81] | Statistical aggregation without pre-specified biological models [82] | Multiplicity-adjusted evidence weights (MAEW) method [81] |
| Nonlinear Effects Capture | Limited to specific modeled interactions | Limited in standard PRS; emerging deep-learning approaches show minimal improvement over linear models [83] | Neural network analysis of 28 UK Biobank traits [83] |
Experimental Protocols for Model Implementation
Polygenic Risk Score Analysis Workflow
The standard protocol for polygenic risk score analysis involves a two-data-set approach requiring both base (GWAS summary statistics) and target (individual genotype and phenotype) data [82]. The critical steps include:
Quality Control Procedures: Base data must undergo heritability check (h²snp > 0.05 recommended) and effect allele verification [82]. Target data requires standard GWAS QC: genotyping rate > 0.99, sample missingness < 0.02, heterozygosity P > 1×10⁻⁶, minor allele frequency > 1%, and imputation info score > 0.8 [82]. For both data sets, file integrity must be verified after transfer to prevent corruption-related artifacts [82].
PRS Calculation and Optimization: PRS are computed as the sum of risk alleles weighted by effect size estimates from GWAS summary statistics [82]. The fundamental formula is:
[ PRSi = \sum{j=1}^{M} \betaj \times G{ij} ]
Where (PRSi) is the polygenic score for individual (i), (\betaj) is the effect size of SNP (j) from base data, and (G_{ij}) is the genotype of individual (i) at SNP (j) [82]. Optimal SNP selection and effect size shrinkage parameters are determined through statistical techniques such as clumping and thresholding or Bayesian approaches [82].
Functional GWAS (fGWAS) for Longitudinal Data
For dynamic traits that evolve over time, the functional GWAS (fGWAS) approach incorporates mathematical functions to model biological processes [84]. The statistical model for longitudinal phenotypic values is expressed as:
[ yi(t{i\tau}) = \sum{j=1}^3 \xii \muj(t{i\tau}) + \beta^T(t{i\tau}) xi + ei(t{i\tau}) + \varepsiloni(t{i\tau}) ]
Where (yi(t{i\tau})) is the trait value for subject (i) at time (t{i\tau}), (\muj(t{i\tau})) is the time-dependent mean value for genotype (j), (\beta) contains time-varying regression coefficients for covariates (xi), and (ei(t{i\tau})) and (\varepsiloni(t{i\tau})) are permanent and random errors, respectively [84].
This approach enables estimation of time-varying additive ((a(t))) and dominant ((d(t))) effects:
[ a(t) = \frac{1}{2}[\mu1(t) - \mu3(t)] ] [ d(t) = \mu2(t) - \frac{1}{2}[\mu1(t) + \mu_3(t)] ]
The fGWAS methodology capitalizes on cumulative phenotypic variation across timepoints, increasing power for gene detection compared to single-timepoint analyses [84].
Biological Model Selection Using MAEW
For selecting appropriate genetic models in association studies, the Multiplicity-Adjusted Evidence Weights (MAEW) method provides empirical selection among five candidate models: null, additive, dominant, recessive, and co-dominant [81]. The protocol involves:
- Calculation of evidence weights based on Bayesian Information Criterion (BIC) for each genetic model [81]
- Multiplicity adjustment through empirical Bayesian probability estimation to control false discovery rate [81]
- Model selection by identifying the model with the highest evidence weight for each variant [81]
This approach enhances biological interpretation and increases validation power in two-stage discovery-validation studies by selecting the most powerful statistical test for follow-up analysis [81].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Research Reagents and Analytical Tools for Genetic Models
| Tool/Reagent | Primary Function | Application Context | Considerations |
|---|---|---|---|
| Array-CGH | Genome-wide copy number variation detection | Identification of structural variants in POI [45] | Resolution ~60 kb; identifies pathogenic CNVs in ~3.6% of POI cases [45] |
| Custom NGS Panels | Targeted sequencing of candidate genes | Mutation detection in known POI genes (e.g., 163-gene panel) [45] | Yield: 28.6% causal SNVs/indels in POI; requires careful variant interpretation [45] |
| LD Score Regression | Heritability estimation from GWAS summary statistics | Quantifying polygenic contribution [80] [82] | Estimates common variant heritability; depends on LD reference panels [80] |
| Burden Heritability Regression (BHR) | Rare variant heritability estimation | Quantifying rare coding variant contributions [80] | Specifically designed for rare variants (MAF < 0.001); accounts for population stratification [80] |
| Neural Network Models | Detection of non-linear genetic effects | Exploring GxG and GxE interactions [83] | Currently shows minimal improvement over linear models for PGS [83] |
| PLINK | Whole-genome association analysis | Standard QC and association testing [82] | Essential for data processing; implements various PRS methods [82] |
The investigation of complex conditions like Premature Ovarian Insufficiency necessitates a multifaceted approach that integrates monogenic, oligogenic, and polygenic perspectives. Current evidence suggests that approximately 57% of idiopathic POI cases harbor identifiable genetic anomalies when combining array-CGH and NGS approaches [45], yet a substantial proportion of genetic liability remains unaccounted for by these methods alone.
The emerging paradigm recognizes that complex traits are influenced by both rare large-effect variants and common small-effect variants acting concurrently [80]. For conditions like POI, this implies that future research strategies should incorporate both targeted sequencing of established candidate genes and genome-wide polygenic approaches to capture the full spectrum of genetic liability. As sample sizes increase and methods refine, the integration of these approaches promises to resolve a greater proportion of the missing heritability that currently limits clinical application.
The field is moving toward models that acknowledge the omnigenic nature of complex traits, where core disease-related genes are embedded in extensive regulatory networks [77]. This perspective, combined with advanced analytical methods that can accommodate both general and specific genetic effects, will ultimately enhance our ability to predict, diagnose, and intervene in complex conditions like POI across diverse populations.
In scientific research, particularly in complex fields like candidate gene studies, the failure to replicate a published finding often triggers an immediate and contentious debate. Is the failure due to a lack of competence on the part of the replicating researchers, or does it stem from fundamental methodological flaws in the original study? This guide objectively compares these two perspectives, arguing that the prevailing overemphasis on researcher competence is not only often inaccurate but also counterproductive to scientific progress. By examining empirical evidence and detailing rigorous experimental protocols, we will demonstrate that methodological integrity, not perceived investigator skill, is the paramount factor in ensuring reliable and replicable research, especially within the context of candidate gene research and replication studies.
The Competence Fallacy: Separating Fact from Fiction
The assumption that a failed replication is the replicator's "fault" is a common but poorly supported intuition. Empirical evidence increasingly shows that this viewpoint is a misconception that fails to withstand scientific scrutiny.
The Empirical Case Against the "Competence Defense"
A direct examination of the relationship between a researcher's established skill and their success in replicating findings provides compelling evidence. An analysis of four Registered Replication Reports (RRRs)—where multiple labs independently conduct the same study—found no consistent relationship between a Principal Investigator's research impact (as measured by h-index) and the success of their replication attempt. This analysis, which included enough data to rule out even very weak relationships, demonstrates that factors other than presumed individual competence are at play in determining replication outcomes [85].
The logical argument against overemphasizing competence is equally important. As noted in critiques of this viewpoint, the scientific process itself is designed with safeguards against individual error. The use of positive and negative controls are time-honored tools that help rule out "bungling" for both expected and unexpected findings. Furthermore, an equal number of practical mistakes can lead to a false positive finding. Holding replication studies to a different, harsher standard of evidence than original studies represents a departure from genuine scientific inquiry [85].
Methodological Flaws: The True Adversary of Replication
While questions of competence often dominate discussions, methodological issues present a far more substantiated and pervasive challenge to replication, particularly in genetically complex fields.
Fundamental Problems in Candidate Gene Research
The candidate gene approach, which focuses on specific genes hypothesized to be involved in a trait or disorder, has been plagued by methodological weaknesses that directly contribute to the replication crisis. The table below summarizes the core methodological flaws identified in the literature.
Table: Key Methodological Flaws in Candidate Gene Research
| Flaw Category | Specific Problem | Impact on Research |
|---|---|---|
| Incomplete Gene Coverage | Investigating only one or a few polymorphisms (SNPs) in a gene rather than capturing its full variation [86]. | Fails to account for other mutations within the same gene that may counteract or enhance the effect of the one studied, leading to false negatives or inaccurate effect sizes. |
| Inadequate Statistical Power | Reliance on sample sizes orders of magnitude too small to detect the small effect sizes typical of complex behavioral traits [87]. | Dramatically increases false positive rates and ensures that even true effects are unlikely to be detected, making replication a near impossibility. |
| Analytical Flexibility & Bias | Employing inadequate analytic procedures, failing to control for higher-order interactions, and not accounting for non-linear relationships [87]. | Increases the risk of false positives and makes results highly sensitive to analytical choices rather than true biological signals. |
| Low Prior Probability of Hypotheses | Focusing on a handful of historically studied polymorphisms without strong, contemporary biological justification [87]. | The starting premise of many studies is weak; most "historical" candidate polymorphisms show no evidence of association in large, well-powered genome-wide studies. |
These flaws create a perfect storm for non-replication. Incomplete gene coverage means the true genetic signal is often missed [86]. Combined with low power, this almost guarantees that the few "significant" findings that are published are likely to be false positives, a problem exacerbated by publication bias that favors the submission and acceptance of positive over null results [87].
The Critical Role of Methodology in Replication Studies
Just as methodological flaws can undermine original studies, they can also doom replication efforts if not carefully addressed. A successful replication requires meticulous planning and design long before data collection begins. Key steps include:
- Preregistration: Publicly sharing the research plan—including hypotheses, design, measures, and analysis plan—before the study is conducted. This increases transparency and reduces bias, allowing others to evaluate the plan and preventing later analytical flexibility [88].
- Power Analysis: Ensuring the replication study has a sufficient sample size to detect the effect of interest with a high degree of probability, directly countering the power problem common in the original research [87].
- Open Communication: Engaging with the original authors to clarify methodologies and request materials, which helps ensure the replication is as accurate as possible [88].
Experimental Protocols for Robust Research
To move beyond the competence debate, the field must adopt more rigorous and standardized experimental protocols. The following section outlines methodologies for both a replication study and a genome-wide approach, which avoids the pitfalls of the candidate gene strategy.
Protocol 1: Conducting a Rigorous Replication Study
This protocol is adapted from guidelines for designing undergraduate replication projects and can be applied to any replication context, including candidate gene research [88].
- Study Selection and Scoping: Choose an original study that is appropriate for your resources and expertise. Evaluate the methodological and theoretical impact of the finding. For a candidate gene study, pay close attention to the gene coverage and statistical power of the original work.
- Preregistration and Transparency:
- Develop a detailed replication plan that exactly specifies the methodology of the original study.
- Preregister this plan on a platform like the Open Science Framework (OSF) or a specialized registry like ClinicalTrials.gov for clinical trials. The preregistration must include the hypothesis, primary and secondary outcome measures, sample size and justification (power analysis), and the exact statistical analysis plan.
- Material Validation and Pilot Testing:
- Obtain all original materials (e.g., survey instruments, genotyping protocols) from the original authors. If unavailable, document any adaptations meticulously.
- Conduct pilot testing of the entire procedure to ensure feasibility and that the methodology produces interpretable data.
- Data Collection and Blindness:
- Implement a double-blind design where possible, so that neither the participants nor the experimenters directly involved in data collection are aware of the study's specific hypotheses or whether it is a replication.
- Use positive controls within the experiment to verify that the methods are capable of detecting an expected effect [85].
- Data Analysis and Reporting:
- Adhere strictly to the preregistered analysis plan.
- Report all results, regardless of outcome, and submit the findings for publication, even if the replication fails. Share data and analysis code publicly to enable further scrutiny.
Replication Study Workflow
Protocol 2: A Modern Genome-Wide Interaction Study
This protocol outlines a methodologically superior alternative to the conventional candidate gene-by-environment (cGxE) interaction study [87].
- Hypothesis and Design:
- Hypothesis: To identify any genetic variants across the entire genome that interact with a specific, well-measured environmental factor to influence a trait of interest.
- Design: A genome-wide association study (GWAS) focused on interaction effects (GxE GWAS), with no a priori restriction to specific candidate genes.
- Sample Collection and Genotyping:
- Collect a very large sample size (typically tens or hundreds of thousands of individuals) to achieve sufficient power to detect interactions of small effect.
- Genotype participants using a high-density genome-wide array, followed by imputation to a reference panel to gain data on millions of common single nucleotide polymorphisms (SNPs).
- Quality Control (QC) and Imputation:
- Perform stringent QC on both genotype and environmental data. This includes filtering out individuals and SNPs with high missing rates, checking for sex discrepancies, and assessing population stratification.
- Impute ungenotyped SNPs using a reference panel (e.g., from the 1000 Genomes Project) to create a uniform set of genetic variants for analysis.
- Statistical Analysis:
- For each SNP, test for an interaction effect between the SNP (coded additively) and the environmental variable on the outcome trait. This is typically done using a linear or logistic regression model that includes the SNP, environment, and SNP-environment interaction term, while adjusting for covariates like age, sex, and genetic principal components to control for population structure.
- Correct for multiple testing across the entire genome using a standard genome-wide significance threshold (e.g., p < 5 × 10^-8).
- Replication and Interpretation:
- Seek to replicate any significant interaction effects in an independent cohort.
- Interpret significant findings in the context of biological pathways and functional genomic data.
GxE GWAS Analysis Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Table: Key Reagents and Resources for Genetic Replication and GxE Studies
| Item | Function/Brief Explanation | Example/Note |
|---|---|---|
| High-Density Genotyping Array | Allows for the simultaneous assaying of hundreds of thousands to millions of genetic variants across the genome. | Foundation for GWAS and GxE GWAS. Examples include Illumina Global Screening Array or Infinium Omni5. |
| Whole Genome Sequencing (WGS) | Provides a complete read of an individual's DNA sequence, capturing virtually all genetic variation. | The gold standard for variant discovery, though more costly than arrays for very large samples. |
| Quality Control (QC) Pipelines | Software tools to filter out low-quality genetic data and individuals, ensuring data integrity before analysis. | Essential for preventing technical artifacts. Examples: PLINK, QCTOOL. |
| Genotype Imputation Server | A computational tool that uses reference haplotypes to infer missing genotypes, increasing the number of testable variants. | Critical for harmonizing data across different arrays and studies. Examples: Michigan Imputation Server, TOPMed Imputation Server. |
| Phenotype & Environment Measure | A validated, reliable tool for assessing the non-genetic factor of interest (e.g., structured clinical interview, environmental exposure assay). | The quality of the "E" in GxE is as important as the "G". Poor measurement dooms the study. |
| Statistical Genetics Software | Programs for performing association and interaction analyses on large-scale genetic data. | Examples: SAIGE, REGENIE, PLINK. |
| Bioinformatics Databases | Repositories of functional genomic data to help interpret the biological significance of identified genetic variants. | Examples: GTEx Portal (for gene expression), ENCODE, GWAS Catalog. |
The evidence clearly demonstrates that the scientific community must shift its focus from questioning researcher competence to rigorously addressing methodological flaws. The empirical data shows no link between a scientist's pedigree and replication success, while the literature is replete with examples of how inadequate gene coverage, underpowered designs, and flexible analyses systematically undermine the reliability of research, particularly in candidate gene studies. By embracing rigorous experimental protocols—including preregistration, large-scale collaborative sampling, and genome-wide approaches—researchers can build a more cumulative, reliable, and trustworthy science. The path forward lies not in ad hominem debates, but in a collective commitment to methodological rigor.
Assessing Success and Integrating Evidence from Multiple Approaches
Establishing a Statistical Framework for Replication Success
In the field of genomic research, particularly in the identification and validation of candidate genes for drug development, the establishment of a robust statistical framework for replication success is paramount. Replication studies serve as the cornerstone for distinguishing true biological signals from false discoveries, ensuring that findings from initial genome-wide association studies (GWAS) and other genomic investigations can be reliably translated into therapeutic targets. The complex nature of biological systems, combined with the challenges of multiple hypothesis testing in high-dimensional data, necessitates a rigorous approach to replication that goes beyond simple statistical significance.
Recent advances in multi-omics technologies and large-scale biobanks have dramatically increased the scope and scale of genomic discovery, making the need for formal replication frameworks more pressing than ever. This guide examines the current methodologies, protocols, and materials essential for establishing such a framework, with a specific focus on candidate genes in drug development contexts. By objectively comparing different analytical approaches and their supporting experimental data, we provide researchers with practical tools to enhance the reliability and success of their replication efforts, ultimately contributing to more efficient drug development pipelines and precision medicine initiatives.
Comparative Analysis of Replication Frameworks and Their Performance
Statistical Foundations for Replication Success
| Framework Component | Traditional GWAS Approach | Integrated Multi-Layer Framework | Performance Metrics |
|---|---|---|---|
| Primary Evidence | Genome-wide significance (P < 5 × 10-8) | Combined P-value from multiple evidence layers [89] | Increased odds ratio for replicated findings |
| Genetic Instruments | Single lead SNPs from discovery GWAS | Cis-eQTLs (within 1Mb of gene), clumped (r2 < 0.1) [89] | F-statistic > 10 for instrument strength |
| Causal Inference | Observational association | Mendelian Randomization with sensitivity analyses [89] | Consistent effect direction across methods |
| Colocalization | Not routinely applied | Bayesian colocalization to confirm shared causal variants [89] | Posterior probability > 80% for shared variant |
| Cross-Population Validation | Sometimes included in large consortia | Multiple independent datasets (e.g., FinnGen, biobanks) [89] | Consistent effect size and direction |
Experimental Validation Platforms
| Validation Method | Throughput | Key Strengths | Key Limitations | Successful Applications |
|---|---|---|---|---|
| CRISPRi Screening | High (100+ genes simultaneously) | Measures genetic interactions; models hypomorphic mutations [90] | Requires specialized cell engineering | SPIDR library identified WDR48-USP1 synthetic lethality [90] |
| Single-Cell Multiomics | Medium (10-100 cells) | Correlates replication timing with gene expression in same cell [91] | Technically challenging; lower throughput | Revealed RT-transcription relationship in mouse embryos [91] |
| Bulk RNA Sequencing | High | Standardized analysis; well-established protocols | Masks cellular heterogeneity | Confirmed differential expression of candidate genes [89] |
| Immunohistochemistry | Low | Spatial context in tissue; protein-level validation | Semi-quantitative; antibody-dependent | Validated protein expression of TNFSF14 in CRC tissues [89] |
Detailed Experimental Protocols for Key Replication Methodologies
Protocol 1: CRISPRi Dual-Guide Screening for Genetic Interactions
Purpose: To comprehensively map genetic interactions and synthetic lethality among DNA damage response genes and candidate genes, providing functional validation for statistical associations [90].
Workflow Steps:
- Library Design: Select at least two sgRNAs per target gene based on the human CRISPRi-v2 library. Include mismatched variant sgRNAs for essential genes to achieve partial knockdown rather than complete knockout [90].
- Vector Cloning: Clone synthesized oligonucleotides into a dual-sgRNA lentiviral expression vector, ensuring each sgRNA combination is uniquely represented [90].
- Cell Line Engineering: Generate clonal cell lines stably expressing catalytically inactive Cas9 fused to a KRAB transcriptional repressor domain [90].
- Viral Transduction: Transduce cells with the lentiviral library at appropriate multiplicity of infection (MOI) to ensure single-copy integration.
- Phenotypic Selection: Collect baseline cells (T0) 96 hours post-transduction and final time point (T14) after 14 days of proliferation [90].
- Sequencing & Analysis: Quantify sgRNA abundance by next-generation sequencing, using specialized pipelines (e.g., GEMINI) to identify genetic interactions exceeding single-gene effects [90].
Quality Control Measures:
- Include non-targeting sgRNA pairs as negative controls
- Ensure equal performance of guide RNAs in either position
- Confirm strong depletion of perfectly matched sgRNAs targeting essential genes
- Compare screen replicates to assess reproducibility [90]
Protocol 2: Integrated Genomic Framework for Causal Gene Prioritization
Purpose: To prioritize causal and druggable genes by integrating Mendelian randomization (MR) with colocalization analysis and functional genomics data [89].
Workflow Steps:
- Druggable Gene Compilation: Curate genes encoding proteins amenable to modulation by small molecules or biologics from databases like DGIdb (e.g., 4,479 unique druggable genes) [89].
- cis-eQTL Acquisition: Obtain cis-expression quantitative trait loci from consortia (e.g., eQTLGen), focusing on variants within 1Mb of transcription start sites with MAF > 0.01 [89].
- Instrument Selection: Extract cis-eQTL SNPs with genome-wide significance (P < 5.0 × 10-8), clump to eliminate LD (r2 < 0.1, window=10,000kb), and calculate F-statistic to assess instrument strength [89].
- Mendelian Randomization: Perform two-sample MR using GWAS summary statistics for the disease of interest, applying sensitivity analyses (e.g., MR-Egger, MR-PRESSO) to assess pleiotropy [89].
- Colocalization Analysis: Apply Bayesian colocalization methods to determine if GWAS and eQTL signals share a common causal variant.
- Replication: Test significant associations in independent datasets (e.g., FinnGen, biobank studies) and perform subtype-stratified analyses [89].
Validation Steps:
- Conduct phenome-wide association studies (PheWAS) to assess off-target effects
- Analyze gene expression patterns in tumor microenvironment using single-cell and bulk RNA-seq [89]
- Perform experimental validation through RT-qPCR and immunohistochemistry in patient samples [89]
The Scientist's Toolkit: Essential Research Reagent Solutions
Core Reagents for Genomic Replication Studies
| Reagent/Category | Specific Examples | Function & Application | Key Considerations |
|---|---|---|---|
| CRISPR Screening Libraries | SPIDR (Systematic Profiling of Interactions in DNA Repair) library [90] | Comprehensive mapping of genetic interactions; contains 697,233 guide-level interactions targeting 548 DDR genes | Includes mismatched sgRNAs for essential genes; enables partial knockdown |
| Druggable Gene Databases | DGIdb v4.2.0, DrugBank [89] | Curated lists of genes encoding proteins amenable to pharmacological modulation | Regular updates essential; includes both clinical and exploratory candidates |
| eQTL Resources | eQTLGen Consortium (31,684 individuals) [89] | Provides large-scale cis-eQTL information for instrument selection in MR studies | Sample size impacts power; ancestry matching with outcome data critical |
| Validation Antibodies | Target-specific validated antibodies (e.g., for TNFSF14, TFRC) [89] | Immunohistochemical validation of protein expression in patient samples | Specificity validation essential; optimal dilution determined empirically |
| scRNA-seq Platforms | 10x Genomics, in-house sc-multiomics protocols [91] | Simultaneous analysis of replication timing and gene expression in individual cells | Compatible with low cell numbers (as few as 10 cells); preserves gDNA and mRNA |
| Statistical Software | R, Python, GEMINI pipeline [90] | Specialized analysis of genetic interactions from CRISPR screening data | Open-source alternatives available; custom scripting often required |
Data Visualization and Interpretation in Replication Studies
Advanced Analytical Approaches for Replication Data
Quantitative Analysis Methods: The interpretation of replication studies relies heavily on robust quantitative data analysis methods, which systematically transform numerical data into meaningful insights using mathematical, statistical, and computational techniques [92]. Descriptive statistics provide the initial snapshot of data characteristics through measures of central tendency (mean, median, mode) and dispersion (range, variance, standard deviation) [92]. Inferential statistics then enable researchers to make generalizations from sample data to broader populations through hypothesis testing, t-tests, ANOVA, and regression analysis [92].
Multi-Layered Evidence Integration: Successful replication frameworks increasingly employ multi-layered analytical approaches that integrate evidence across different data types and experimental modalities. For example, the combination of Mendelian randomization with colocalization analysis and functional genomic validation provides complementary evidence strands that collectively strengthen causal inference [89]. This integrated approach moves beyond traditional single-metric significance testing to create a comprehensive evidence base for replication success.
The establishment of a statistical framework for replication success requires integration of multiple evidence layers, from robust statistical genetics and causal inference methods to functional validation in biologically relevant systems. The comparative analysis presented in this guide demonstrates that multi-layer approaches—combining Mendelian randomization, colocalization, cross-population validation, and experimental assessment—consistently outperform single-method frameworks in identifying replicable candidate genes for drug development.
As genomic technologies continue to evolve, the most successful replication frameworks will be those that adapt to incorporate new data types while maintaining rigorous statistical standards. The protocols, reagents, and analytical approaches detailed here provide researchers with practical tools to enhance the reliability of their replication efforts, ultimately contributing to more efficient translation of genomic discoveries into clinical applications. Future directions will likely include greater incorporation of single-cell multiomics data, more sophisticated computational models for integrating diverse evidence types, and standardized frameworks for assessing replication success across diverse populations.
The advent of genome-wide association studies (GWAS) has revolutionized the identification of genetic variants associated with complex diseases and traits. This approach has been particularly impactful in the field of human reproductive health and livestock breeding, where it has enabled the large-scale discovery of candidate genes without requiring prior knowledge of biological function. In the context of premature ovarian insufficiency (POI), GWAS has promised to unravel the complex genetic architecture underlying this clinically heterogeneous condition, which affects approximately 1-3% of women under 40 and is characterized by elevated follicle-stimulating hormone (FSH) levels and diminished ovarian reserve [93] [94].
Despite the initial enthusiasm surrounding GWAS discoveries, the field now faces a critical challenge: many candidate genes identified through GWAS fail to replicate in subsequent studies or lack functional validation. This replication crisis stems from multiple factors, including insufficient statistical power, population stratification, phenotypic heterogeneity, and inadequate correction for multiple testing. Furthermore, the transition from statistical association to biological mechanism remains a significant bottleneck. As we move further into the GWAS era, a systematic evaluation of past claims is essential to distinguish robust genetic associations from false positives and to refine our understanding of genuine biological pathways.
This review employs a systematic framework to evaluate candidate genes in POI research, with a particular focus on the integration of multi-omics data and functional validation. We compare the performance of various GWAS approaches across different populations and species, analyze the experimental evidence supporting prominent candidate genes, and provide standardized protocols for gene validation. By critically appraising the current state of the field, we aim to establish best practices for future research and facilitate the translation of GWAS findings into clinical applications.
Evaluating GWAS Methodologies Across Studies
The credibility of candidate genes hinges fundamentally on the methodological rigor of the GWAS from which they emerge. Significant variability in study design, statistical power, and validation strategies across experiments directly impacts the reproducibility of their findings. The table below systematically compares the key methodological aspects of recent GWAS that have investigated reproductive traits across different species and populations:
Table 1: Methodological Comparison of GWAS on Reproductive Traits
| Study Focus | Population/Species | Sample Size | Key Candidate Genes Identified | Validation Approach | Heritability (h²) |
|---|---|---|---|---|---|
| Primary Ovarian Insufficiency [93] | Human (40 bPOI patients, 47 controls) | 87 | YBX1 | In vitro functional assays (knockdown/overexpression) in KGN cell line | Not reported |
| Dairy Cattle Reproduction [95] | Holstein Bulls (50,309) | 50,309 | AOPEP, GC, E2F6, MGST1 | Bayesian fine-mapping | Varies by trait |
| Chicken Body Weight [28] | Three chicken populations (1,143 total) | 1,143 | KPNA3, CAB39L | Multi-population meta-analysis | 0.21 - 0.48 for BW traits |
| Pig Reproduction [96] | Iranian Holstein Cows (number varied by trait) | 50K SNP chip | ATG7, PTPN5 (heifers); LPL, SERP2 (cows) | Gene ontology analysis | Low (e.g., 0.091 for teat number) |
| Pig Loin Muscle Area [97] | Three pig breeds (4,175 total) | 4,175 | NDUFS4, ARL15, FST, ADAM12 | Meta-analysis, QTL comparison | 0.40 - 0.63 |
| Sheep Reproduction & Growth [98] | Tianmu Prolific Sheep (483 ewes) | 483 | DEPTOR, GNG12, WWOX, INHA | GO and KEGG enrichment analysis | Not reported |
The comparative analysis reveals critical methodological insights. First, sample size remains a fundamental differentiator. The dairy cattle study [95], with over 50,000 individuals, exemplifies the power of large-scale sequencing to detect significant association peaks, including 126 novel findings. In contrast, the human POI study [93] utilized a modest sample of 87 participants, which increases the risk of false positives and limits the detection of variants with small effect sizes.
Second, the approach to validation varies significantly. The most direct functional validation was performed in the POI study, where YBX1's role was confirmed through in vitro experiments [93]. Conversely, agricultural studies often rely on meta-analysis [28] [97] and statistical fine-mapping [95] to boost power and refine candidate regions. These approaches enhance confidence in associations but do not constitute biological validation.
Third, population genetic structure is handled differently across studies. The chicken body weight study [28] explicitly addressed genetic heterogeneity by analyzing three distinct populations and using meta-analysis to identify robust cross-population signals. This approach helps distinguish genuine causal variants from population-specific associations.
The technical platform also influences outcomes. While earlier studies relied on mid-density SNP chips (e.g., 50K in pigs [99]), the trend is shifting toward whole-genome sequencing [98] or imputation to sequence-level [95] [97], which provides more complete genomic coverage and improves the detection of causal variants.
The Replication Crisis: A Case Study of POI Candidate Genes
Premature ovarian insufficiency represents a compelling case study of the challenges in candidate gene replication. The condition's heterogeneity, combined with typically small sample sizes in human studies, has resulted in numerous proposed candidate genes with inconsistent validation across populations.
The YBX1 gene exemplifies both the potential and pitfalls of GWAS candidates. A 2025 study demonstrated that YBX1 expression is decreased in granulosa cells from POI patients and regulates cell cycle progression in an m5C-dependent manner [93]. This finding is mechanistically insightful, as it links RNA epigenetics with granulosa cell dysfunction. However, independent replication of this specific association is still lacking, and the sample size (87 participants) necessitates validation in larger cohorts.
Beyond YBX1, the table below compares previously proposed POI candidate genes against current evidence:
Table 2: Evaluation of Candidate Genes in Premature Ovarian Insufficiency
| Candidate Gene | Original Proposed Function | Supporting Evidence Level | Replication Status | Known Mechanisms |
|---|---|---|---|---|
| YBX1 [93] | m5C reader regulating granulosa cell cycle | Functional evidence in cell lines | Initial discovery, needs independent replication | Stabilizes cell cycle-associated transcripts via m5C reading |
| BMP15 | Oocyte-secreted factor regulating folliculogenesis | Multiple association studies | Partially replicated, population-specific effects | Member of TGF-β superfamily, promotes follicular development |
| FMR1 (premutation) | RNA toxicity affecting ovarian reserve | Strong clinical evidence | Well-replicated in specific POI subpopulation | Trinucleotide repeat expansion causing mRNA toxicity |
| FOXO3a | Regulation of primordial follicle activation | Animal model evidence | Inconsistent in human populations | Transcription factor involved in stress response pathways |
| EIF4ENIF1 | Mitochondrial function in oocytes | Familial cases | Limited replication | Nucleocytoplasmic transport, implicated in mitochondrial dysfunction |
The replication challenges in POI genetics stem from several sources. Clinical heterogeneity is paramount, as POI can result from genetic, autoimmune, iatrogenic, or idiopathic causes [94]. Without careful sub-phenotyping, genetic studies may combine etiologically distinct patients, diluting true signals. Additionally, incomplete penetrance and polygenic inheritance complicate the identification of monogenic causes.
The emergence of new biological mechanisms adds another layer of complexity. The YBX1 study [93] highlights the growing importance of RNA modifications in POI pathogenesis, particularly m5C methylation. This represents an epigenetic dimension beyond traditional protein-coding mutations that may explain additional cases but requires specialized methodologies to detect.
Furthermore, species differences in reproductive biology limit the direct translation of findings from model organisms to humans. While genes like FOXO3a show strong effects in mice, their role in human POI remains less clear. This discrepancy underscores the need for human cell line models, such as the KGN granulosa cell line used in YBX1 validation [93].
Experimental Protocols for Candidate Gene Validation
The transition from statistical association to biological mechanism requires a systematic validation pipeline. Below, we outline standardized experimental protocols for confirming the functional role of candidate genes identified through GWAS, with examples from recent POI research.
In Vitro Functional Validation in Granulosa Cell Models
The KGN human granulosa cell line has emerged as a valuable model for studying POI pathogenesis in vitro [93]. The following protocol details the key steps for functional validation:
Cell Culture and Transfection:
- Culture KGN cells in high-glucose DMEM supplemented with 5% fetal bovine serum and 1% penicillin-streptomycin at 37°C with 5% CO₂ [93].
- At 70-80% confluence, transfect cells with 25 nM siRNA targeting the candidate gene or negative control using Lipofectamine RNAiMAX in Opti-MEM [93].
- For overexpression studies, transfert cells with plasmid constructs (e.g., pCMV3-HA-tagged gene) using Lipofectamine 3000 [93].
- Harvest cells 48 hours post-transfection for downstream analysis.
Phenotypic Assays:
- Cell Proliferation Analysis: Assess using CCK-8 assays or EdU incorporation to determine if candidate gene perturbation affects granulosa cell growth [93].
- Cell Cycle Profiling: Analyze cell cycle distribution via flow cytometry after propidium iodide staining. YBX1 knockdown, for instance, prevented G1 to S phase transition [93].
- Gene Expression Analysis: Extract total RNA using RNAiso Plus kit and reverse transcribe to cDNA. Perform quantitative real-time PCR with ChamQ Universal SYBR qPCR Master Mix to examine expression of cell cycle-associated genes [93].
Multi-Omics Integration Protocol
Integrating genomic data with transcriptomic and epigenomic information strengthens causal inference for GWAS candidates:
Transcriptome-Wide Association Study (TWAS):
- Extract RNA from relevant tissues (e.g., granulosa cells) and perform RNA-seq on Illumina platforms [100].
- Map clean reads to the reference genome using Hisat2 and quantify gene expression levels as FPKM [100].
- Conduct association analysis between expression levels and phenotypic traits using linear mixed models in software such as GEMMA [100].
Epigenomic Analysis:
- Perform methylated RNA immunoprecipitation sequencing (MeRIP-seq) to map m5C or m6A modifications [93].
- Integrate methylome data with transcriptome profiles to identify genes whose stability may be regulated by RNA modifications.
- Use chromatin immunoprecipitation (ChIP) for transcription factors or histone modifications to link genetic variants to regulatory elements.
In Vivo Validation Considerations
While human studies are ultimately required, model organisms provide important preliminary evidence:
- Generate tissue-specific knockout mice to recapitulate human POI phenotypes.
- Monitor estrous cycles, measure serum FSH/LH levels, and perform histological analysis of ovarian sections.
- Assess fertility through timed mating trials and count litter sizes.
These experimental workflows can be visualized in the following diagram:
Figure 1: Candidate Gene Validation Pipeline. This workflow outlines the systematic process from initial GWAS discovery to clinical application, emphasizing the critical role of functional and mechanistic studies.
Essential Research Reagents and Tools
Robust validation of candidate genes requires high-quality research reagents and standardized experimental tools. The following table catalogues essential resources for studying POI candidate genes, with examples from recent publications:
Table 3: Essential Research Reagent Solutions for Candidate Gene Validation
| Reagent/Tool | Specification | Research Application | Example Use |
|---|---|---|---|
| KGN Cell Line [93] | Immortalized human granulosa cell line | In vitro modeling of granulosa cell biology | Studying YBX1 effects on cell cycle progression [93] |
| siRNA/shRNA | 25 nM targeting sequence | Gene knockdown studies | YBX1 silencing to assess proliferation effects [93] |
| Expression Plasmids | pCMV3-HA-tagged | Candidate gene overexpression | YBX1 overexpression to confirm proliferation role [93] |
| RNA Extraction Kit | RNAiso Plus | High-quality RNA isolation | Preparing samples for transcriptome analysis [93] |
| qPCR Master Mix | ChamQ Universal SYBR | Gene expression quantification | Validating transcript stability in YBX1 studies [93] |
| GWAS Software | GCTA, GEMMA, PLINK | Association analysis | Identifying significant variant-trait associations [28] [98] |
| Imputation Tools | Beagle 5.1 | Genotype imputation | Enhancing SNP density using reference panels [28] |
| Functional Annotation | VEP, DAVID | Biological context interpretation | GO and KEGG pathway enrichment analysis [99] [98] |
The selection of appropriate research tools significantly impacts the reliability of validation studies. The KGN cell line has become particularly valuable for POI research as it maintains many characteristics of human granulosa cells, including expression of FSH receptors and steroidogenic enzymes [93]. For genetic manipulation, the combination of siRNA-mediated knockdown and cDNA overexpression provides compelling evidence for a gene's functional role, as demonstrated in the YBX1 study where both approaches produced concordant effects on proliferation [93].
Bioinformatics tools for GWAS and functional annotation continue to evolve. Recent studies emphasize the importance of multi-omics integration, combining GWAS with transcriptomic data (TWAS) to prioritize candidate genes [100]. Software such as GCTA for association testing [28] and DAVID for functional annotation [99] have become standard in the field. Additionally, the use of imputation tools like Beagle [28] to enhance SNP coverage from array-based genotyping to sequence-level data has improved the resolution of association signals.
For animal studies, standardized phenotyping protocols are essential. In sheep reproductive studies, for example, precise measurement of body size traits (withers height, chest depth, cannon bone circumference) using standardized measuring sticks and flexible tape measures ensures data consistency [98]. Similarly, in pig research, adjusting traits like loin muscle area to a standard 100 kg body weight enables meaningful comparisons across breeds [97].
Signaling Pathways and Molecular Mechanisms
The integration of GWAS findings with molecular signaling pathways is essential for transforming statistical associations into biological insights. In POI research, several key pathways have emerged as critically involved in ovarian function, with YBX1 representing a recently elucidated mechanism connecting RNA epigenetics with granulosa cell function.
The YBX1-mediated pathway exemplifies how GWAS candidates can reveal novel biological mechanisms in POI:
Figure 2: YBX1-m5C Pathway in Granulosa Cell Dysfunction. This diagram illustrates the molecular mechanism through which aberrant YBX1 downregulation leads to POI pathogenesis via m5C-dependent transcript destabilization.
The pathway illustrates several key biological concepts. First, YBX1 functions as an m5C reader protein that recognizes specific RNA modifications and regulates transcript stability [93]. When YBX1 is downregulated in POI patients, it fails to stabilize critical cell cycle-associated mRNAs, leading to their destabilization. This ultimately impairs the G1 to S phase transition in granulosa cells, reducing proliferation and contributing to ovarian dysfunction [93].
Beyond the YBX1 pathway, other important signaling cascades have been implicated in POI pathogenesis through GWAS and functional studies:
TGF-β Signaling Pathway:
- Members of the TGF-β superfamily, including BMP15 and GDF9, play crucial roles in folliculogenesis
- Mutations in these genes disrupt follicular development and oocyte quality
- This pathway represents one of the best-validated mechanisms in POI genetics
Hormone Signaling Pathways:
- FSH receptor signaling is essential for antral follicle development
- Estrogen receptor mutations can cause follicular maturation defects
- These pathways connect genetic susceptibility with endocrine features of POI
DNA Damage Repair Pathways:
- Genes involved in meiotic recombination and DNA repair contribute to ovarian reserve maintenance
- Mutations in MCM8, MCM9, and other DNA repair genes cause premature follicle depletion
- This mechanism highlights the particular vulnerability of oocytes to genotoxic stress
Mitochondrial Function Pathways:
- Adequate mitochondrial function is crucial for oocyte competence
- Genes involved in oxidative phosphorylation and energy metabolism affect ovarian aging
- This pathway connects metabolic health with reproductive longevity
The convergence of multiple candidate genes onto shared biological pathways strengthens their credibility as genuine POI risk factors. Future studies should prioritize pathway-based analyses rather than focusing exclusively on individual genes.
The systematic evaluation of candidate genes in the GWAS era reveals both significant challenges and promising avenues for advancing POI research. The replication crisis affecting many proposed candidate genes stems from methodological limitations rather than intrinsic flaws in the GW approach itself. Small sample sizes, phenotypic heterogeneity, and insufficient validation have hampered progress in translating statistical associations into biological mechanisms.
Moving forward, several strategies can enhance the reliability of candidate gene identification. First, collaborative consortia must prioritize large-scale meta-analyses to achieve sufficient statistical power. The success of such approaches in agricultural genetics [95] [28] [97] provides a template for human studies. Second, standardized validation pipelines incorporating multi-omics data and functional experiments should become mandatory for claiming novel candidate genes. The YBX1 study [93] offers a exemplary model of this approach, combining association data with mechanistic insights into m5C-dependent regulation.
Third, advanced computational methods including Bayesian fine-mapping [95] and TWAS [100] can prioritize the most likely causal genes from association signals. Finally, cross-species validation using appropriate animal models and human cell lines can bridge the gap between association and function.
For the clinical translation of POI genetics, the most immediate application may be in risk prediction and personalized management rather than novel therapeutics. Polygenic risk scores combining multiple validated variants could identify women at increased risk for early ovarian aging, enabling proactive fertility preservation. Additionally, the identification of subtypes based on genetic markers may lead to more tailored hormonal regimens.
The journey from GWAS signal to biological mechanism remains challenging, but the systematic approach outlined in this review provides a roadmap for distinguishing genuine causal genes from statistical artifacts. As methods continue to evolve and sample sizes increase, the promise of GWAS to unravel the genetic architecture of complex traits like POI will be increasingly realized.
The pursuit of candidate genes for complex traits like hypertension represents a crucial pathway in unraveling the genetic architecture of cardiovascular disease. Successful replication of genetic associations across diverse populations stands as a critical validation step before these findings can transition into clinical applications or drug development pipelines. Within this framework, genes encoding key components of the renin-angiotensin-aldosterone system (RAAS)—particularly angiotensinogen (AGT) and aldosterone synthase (CYP11B2)—have emerged as prominent candidates for essential hypertension.
Despite extensive investigation, literature reveals contradictory associations between these candidate genes and hypertension across different studies, highlighting the challenge of replicating genetic findings in distinct populations. [101] Much of this inconsistency stems from early meta-analyses that failed to account for population genetic ancestry as a confounding factor. [101] This case study examines how rigorous, ancestry-stratifed replication efforts have substantiated the roles of AGT and CYP11B2 polymorphisms in hypertension pathogenesis, providing a model for future candidate gene research in the era of precision medicine.
Methods & Experimental Protocols in Replication Studies
Population Stratification and Inclusion Criteria
Modern replication studies employ stringent methodological frameworks to ensure robust findings. The contemporary approach involves:
- Systematic Literature Retrieval: Comprehensive searches across PubMed, Cochrane, Web of Science, and other databases using structured search strategies with precise Medical Subject Headings (MeSH) terms and keywords. [101] [102]
- Ancestry-Informed Stratification: Categorizing study populations based on genetic ancestry and/or major geographical origins rather than broad racial categories, recognizing that different genotype and allele frequencies in diverse populations result in different genetic associations with hypertension. [101] Key population groups include European, Southern Chinese, East Asian (Northern Chinese and Japanese), Southern Indian, Northern Indian, Latin American, African, and Middle Eastern.
- Standardized Phenotyping: Applying consistent diagnostic criteria for essential hypertension, typically defined as blood pressure ≥140 mmHg systolic or ≥90 mmHg diastolic or use of anti-hypertensive medication, with normotensive controls defined as blood pressure <140/90 mmHg. [101]
Genotyping Methodologies
Reliable genotyping forms the technical foundation of replication studies. Established techniques include:
- Polymerase Chain Reaction Restriction Fragment Length Polymorphism (PCR-RFLP): Conventional method employing specific restriction enzymes (HaeIII for CYP11B2-344T/C) to digest amplified products, followed by electrophoresis on agarose gels stained with ethidium bromide for visualization. [103] [104]
- Standardized Primer Design and Amplification: Using Primer 5.0 software or similar tools to design sequence-specific primers, with amplification reactions typically in 25μL volumes containing PCR Taq Master Mix, upstream and downstream primers, DNA template, and deionized water. [104]
- Quality Control Measures: Implementing stringent quality checks including suspicion of genotyping errors when reported frequencies significantly deviate from expected values or potential strand-flipping issues. [101]
Table 1: Key Research Reagent Solutions for Genotyping Experiments
| Research Reagent | Function/Application | Specific Example |
|---|---|---|
| Restriction Enzymes | Digest PCR products for polymorphism identification | HaeIII for CYP11B2 [103], AluI for ACE2 [104] |
| DNA Extraction Kits | Isolate genomic DNA from blood samples | Whole-blood rapid genomic DNA extraction kits [104] |
| PCR Master Mix | Amplify target gene sequences | Standard Taq-based mixes with optimized buffer conditions [104] |
| Electrophoresis Materials | Separate DNA fragments by size | 2.0-2.5% agarose gels with ethidium bromide staining [103] [104] |
Replication Findings for AGT and CYP11B2
AGT (rs699) Polymorphism Associations
The AGT rs699 polymorphism (resulting in M235T substitution) has been extensively studied across global populations. Recent ancestry-stratified meta-analyses provide compelling evidence for population-specific effects:
- South Asian Populations: Significant association observed among Indian populations for both allele frequency (P=0.03, OR: 1.37, 95% CI: 1.03-1.82) and dominant mode of inheritance (GG+GA vs. AA: P=0.009, OR: 1.45, 95% CI: 1.09-1.91). [101]
- Biological Mechanism: The rs699-G allele is attributed to elevated plasma angiotensinogen levels, enhancing the substrate availability for renin-mediated angiotensin I production and consequently influencing blood pressure regulation through the RAAS pathway. [101]
- Population Contrasts: Unlike the consistent associations observed in South Asians, European and East Asian populations showed more variable associations, underscoring the importance of population genetic background in modifying disease risk. [101]
CYP11B2 (rs1799998) Polymorphism Associations
The CYP11B2 gene, encoding aldosterone synthase, contains a functional polymorphism (T(-344)C, rs1799998) that alters a putative steroidogenic factor-1 binding site, leading to downregulation of CYP11B2 promoter activity and consequent altered gene expression. [101]
- European Populations: Demonstrated significant associations with hypertension across multiple genetic models: allele model (P=6.9×10⁻⁵, OR: 0.82, 95% CI: 0.74-0.9), recessive model (P=6.38×10⁻⁵, OR: 0.7, 95% CI: 0.59-0.83), and dominant model (P=0.008, OR: 0.81, 95% CI: 0.7-0.94). [101]
- East Asian Populations: Early studies in Chinese populations identified significant associations, with the TT genotype and T allele frequency significantly higher in hypertensive patients compared to controls (P<0.05), suggesting the T allele may serve as a genetic marker for hypertension in certain populations. [103]
- Gene-Environment Interactions: Studies among steelworkers demonstrated interactive effects between CYP11B2-344T/C polymorphisms and occupational noise exposure, with the TC genotype amplifying hypertension risk when combined with noise exposure. [104]
Gene-Gene Interactions in Hypertension Pathogenesis
Beyond individual gene effects, epistatic interactions between RAAS genes significantly influence hypertension risk:
- ACE and CYP11B2 Interaction: Carriers of both the ACE I/D DD genotype and CYP11B2-344T/C TC genotype exhibited substantially increased hypertension risk (OR: 1.99, 95% CI: 1.14-3.51 for ACE DD alone), demonstrating multiplicative interaction effects (P=0.041). [104]
- Clinical Subtypes: Genetic effects appear particularly pronounced in specific hypertension subtypes. CYP11B2, REN, and AGT gene combinations differentiate between low-renin hyperaldosteronism and low-renin hypertension with normal aldosterone levels, suggesting distinct genetic architectures for these clinical phenotypes. [105]
Table 2: Summary of Replicated Genetic Associations for AGT and CYP11B2
| Gene/Polymorphism | Risk Allele/Genotype | Associated Population | Effect Size (Odds Ratio) | Significance |
|---|---|---|---|---|
| AGT (rs699) | G allele | South Asian | 1.37 (1.03-1.82) | P=0.03 [101] |
| AGT (rs699) | GG+GA genotype | South Asian | 1.45 (1.09-1.91) | P=0.009 [101] |
| CYP11B2 (rs1799998) | C allele | European | 0.82 (0.74-0.90) | P=6.9×10⁻⁵ [101] |
| CYP11B2 (rs1799998) | CC genotype | European | 0.70 (0.59-0.83) | P=6.38×10⁻⁵ [101] |
| ACE (I/D) + CYP11B2 | DD + TC genotypes | Chinese males | 1.99 (1.14-3.51) | P=0.041 (interaction) [104] |
Biological Pathways and Mechanisms
The biological plausibility of AGT and CYP11B2 associations with hypertension is firmly grounded in their integral roles within the RAAS pathway, which represents a core blood pressure regulatory system.
Diagram 1: RAAS Pathway with Genetic Modulation Points. AGT rs699-G increases angiotensinogen production, while CYP11B2 rs1799998 alters promoter activity, both influencing blood pressure regulation.
AGT serves as the precursor to angiotensin I and is primarily synthesized in the liver. The rs699 polymorphism influences circulating AGT levels, with the G allele associated with elevated plasma angiotensinogen. [101] This increased substrate availability enhances renin-mediated conversion to angiotensin I, subsequently leading to increased angiotensin II production—a potent vasoconstrictor—and ultimately elevated blood pressure.
CYP11B2, expressed in the adrenal zona glomerulosa, catalyzes the final steps of aldosterone synthesis. The rs1799998 polymorphism alters a transcription binding site for steroidogenic factor-1, resulting in downregulated promoter activity and consequent modulation of aldosterone production. [101] Aldosterone promotes sodium reabsorption in the renal tubules, expanding plasma volume and increasing blood pressure.
These genetic variations contribute to intermediate hypertension phenotypes, particularly salt-sensitive hypertension. AGT has been associated with normal renin salt-sensitive hypertension, while CYP11B2 is linked to low renin salt-sensitive hypertension, [101] reflecting distinct pathophysiological mechanisms within the broader hypertension spectrum.
Discussion: Implications for Drug Development and Precision Medicine
Pharmacogenetic Applications
The successful replication of AGT and CYP11B2 associations across populations carries significant implications for antihypertensive drug development and personalized treatment strategies:
- Therapeutic Targeting: CYP11B2 represents a direct therapeutic target for aldosterone synthase inhibition, with compounds like FAD286 and its derivatives showing potent inhibition of cellular aldosterone production (IC50 values as low as 1-2 nM for optimized compounds). [106]
- Pharmacogenetic Stratification: Genotype-informed treatment approaches may optimize therapeutic efficacy; for instance, patients with AGT or CYP11B2 risk genotypes might respond preferentially to RAAS-targeted therapies like ACE inhibitors or mineralocorticoid receptor antagonists. [107]
- Clinical Trial Design: Incorporating genetic stratification into clinical trials may enhance patient selection and improve trial success rates by identifying genetically homogeneous subgroups most likely to benefit from targeted therapies.
Methodological Advances in Replication Studies
This case study highlights several methodological refinements that have strengthened candidate gene replication efforts:
- Ancestry-Informed Meta-Analysis: Contemporary approaches that categorize data by genetic ancestry rather than broad geographical labels have helped resolve previous contradictory findings and established population-specific effect sizes. [101]
- Standardized Phenotyping: Precise intermediate phenotyping, including renin and aldosterone profiling, has revealed genetic associations with specific hypertension subtypes, particularly low-renin forms associated with CYP11B2 variants. [105]
- Gene-Environment Interaction Modeling: Incorporating environmental exposures like occupational noise has elucidated how genetic risk manifests in specific environmental contexts, providing more nuanced risk prediction models. [104]
The successful replication of AGT and CYP11B2 polymorphisms in hypertension across diverse populations represents a milestone in cardiovascular genetics, validating these candidates as bona fide contributors to blood pressure regulation. This replication success story underscores several critical principles for candidate gene research: the necessity of ancestry-informed analyses, the importance of biological plausibility grounded in known physiological pathways, and the value of examining gene-gene and gene-environment interactions.
These findings provide essential insights for selecting appropriate pharmacogenetic markers for effective hypertension management across different populations. [101] As precision medicine approaches continue to evolve, the robust replication of AGT and CYP11B2 associations establishes a foundation for genetically-informed hypertension management strategies that account for individual genetic background, ultimately supporting the transition from one-size-fits-all approaches to personalized therapeutic interventions.
In the field of candidate gene research, replication of initial findings in independent cohorts, while crucial, often fails to bridge the translational "valley of death." This guide compares the foundational, yet limited, approach of replication studies with the target-driven process of functional validation. We demonstrate that while replication confirms statistical association, functional validation provides the biological causality required to advance drug targets, supported by experimental data from genomic and single-cell transcriptomic studies. The comparative analysis underscores that integrating both strategies within a phased framework, enhanced by rigorous prioritization, is essential for de-risking therapeutic development.
The process of identifying and confirming candidate genes for therapeutic development extends far beyond initial discovery. A foundational challenge in biomedical research is the high failure rate of promising preclinical findings when they enter human trials, a problem known as the "valley of death" [108]. While replication—repeating an analysis in a similar, independent population under identical conditions—is a necessary first step for verification, it is often insufficient to justify the immense investment in clinical development [109]. Replication can confirm that a statistical association is not a spurious finding, but it does not demonstrate that the gene plays a causal role in the disease phenotype.
This is where functional validation becomes critical. Functional validation involves direct experimental perturbation of a candidate gene (e.g., via knockdown, knockout, or overexpression) in relevant model systems to assess its impact on biological processes and disease-related phenotypes [110] [111]. It is the process of moving from a statistical correlation to a demonstrated biological function. For researchers and drug development professionals, understanding the distinction, appropriate application, and synergy between these two approaches is paramount for selecting viable therapeutic targets. This guide provides a objective comparison of these methodologies, their outputs, and their collective role in strengthening the path from gene discovery to clinical application.
Defining the Concepts: Replication vs. Functional Validation
A clear understanding of the terminology is essential for designing robust research strategies. The terms "replication" and "validation" are often used interchangeably, but they represent distinct concepts in the research workflow [109].
Replication: A Test of Statistical Robustness
Replication is the more straightforward concept. Its primary goal is to assess whether a previously observed statistical association can be reproduced under identical conditions in an independent but similar study sample. The ideal replication study minimizes systematic differences between the discovery and replication samples; any discrepancy in findings is then presumed to be due to random variation [109].
- Key Characteristics: Independence of samples, similarity in population demographics (sex, age, ethnicity), and identical laboratory procedures, data processing pipelines, and analytical approaches.
- Primary Goal: To confirm that an initial finding is statistically robust and not a false positive.
- In Practice: True replication is challenging, especially in fields like metabolomics where intra-individual variability is high. Many efforts described as "replication" actually fall under the broader umbrella of validation [109].
Validation: Establishing Biological and Functional Relevance
Validation is a broader and more complex term that encompasses several distinct concepts [109]:
- Technical Validation: This established meaning refers to confirming results using a different, often more precise, laboratory technique within the same population. In genomics, this might mean using Sanger sequencing to confirm a variant identified via high-throughput sequencing. In metabolomics, targeted assays are used to validate hits from an untargeted discovery platform [109].
- Functional Validation: This goes beyond technical confirmation to interrogate the biological role of a candidate gene. It involves experimental manipulation to establish a cause-effect relationship between the gene and a phenotype. As exemplified in a study on tip endothelial cells, functional validation uses tools like siRNA knockdown to assess whether a gene influences critical processes like cell migration or sprouting angiogenesis [111].
- Predictive Model Validation: This involves internal validation (e.g., cross-validation, bootstrapping) to test for overfitting in models built to predict disease risk or outcome, and external validation in a different population [109].
Comparative Analysis: Replication versus Functional Validation
The following table provides a structured, point-by-point comparison of these two critical research phases, summarizing their distinct goals, methodologies, and outputs.
Table 1: A direct comparison of replication and functional validation in candidate gene research
| Aspect | Replication | Functional Validation |
|---|---|---|
| Primary Goal | Confirm statistical association and minimize false positives [109] | Establish biological causality and mechanism of action [110] [111] |
| Core Question | "Can we observe this same association again?" | "Does manipulating this gene directly impact the phenotype?" |
| Methodology | Independent population studies with identical protocols and analytics [109] | Experimental perturbation (e.g., RNAi, CRISPR) in model systems; phenotypic assays [110] [111] |
| Key Outcome Measure | Consistency of statistical association (e.g., p-value, effect size) | Measurable change in a biologically relevant phenotype (e.g., migration, viability) [111] |
| Role in Translation | Builds confidence in the initial observation | Provides direct evidence for therapeutic tractability and de-risks downstream investment [108] |
| Limitations | Does not prove causality; prone to heterogeneity between cohorts | Findings in model systems may not always translate to humans; can be costly and time-consuming |
The journey from a list of candidate genes from a single-cell RNA-sequencing (scRNA-seq) study to a validated target involves a critical prioritization step. The torrent of data from scRNA-seq studies produces long, ranked lists of marker genes, but it remains unknown how many are functionally relevant [111]. A key strategy is to use frameworks like the Guidelines On Target Assessment for Innovative Therapeutics (GOT-IT) to prioritize candidates based on criteria such as target-disease linkage, target-related safety, novelty, and technical feasibility before embarking on laborious functional assays [111].
Table 2: Experimental outcomes from a functional validation study of prioritized tip endothelial cell genes
| Candidate Gene | Prioritization Rationale | Experimental Assay | Key Functional Outcome (upon knockdown) | Validation Result |
|---|---|---|---|---|
| CD93 | Enriched in tip cells; limited angiogenesis publications [111] | HUVEC migration & sprouting [111] | Impaired migration; reduced sprouting capacity [111] | Successful |
| ADGRL4 (ELTD1) | Tip cell-specific marker; mechanism unknown [111] | HUVEC migration & sprouting [111] | Impaired migration; reduced sprouting capacity [111] | Successful |
| GJA1 | Involved in gap junctions; minimally characterized in angiogenesis [111] | HUVEC migration & sprouting [111] | Impaired migration; reduced sprouting capacity [111] | Successful |
| CCDC85B | A "mystery gene" with no prior annotation in angiogenesis [111] | HUVEC migration & sprouting [111] | Impaired migration; reduced sprouting capacity [111] | Successful |
| MYH9 | Cytoskeletal role; limited functional data in ECs [111] | HUVEC proliferation & migration [111] | Altered phenotype in functional assays [111] | Successful |
| TCF4 | Transcription factor; poorly described in tip EC context [111] | HUVEC proliferation & migration [111] | No significant impact on proliferation or migration [111] | Unsuccessful |
Experimental Protocols for Functional Validation
A typical workflow for the functional validation of a candidate gene involves a sequence of well-established molecular and cellular biology techniques. The following protocol outlines the key steps, as applied in recent research.
Protocol: In Vitro Functional Validation via Gene Knockdown in Endothelial Cells
This protocol is adapted from methodologies used to validate novel tip endothelial cell genes, such as CD93 and ADGRL4 [111].
1. Gene Prioritization:
- Input: A ranked list of candidate genes from a discovery platform (e.g., GWAS, scRNA-seq).
- Procedure: Apply prioritization criteria. This includes assessing target-disease linkage (e.g., is the gene specific to a key disease cell type?), target-related safety (e.g., excluding genes linked to other diseases), strategic novelty (e.g., focusing on genes with fewer than 20 related publications), and technical feasibility (e.g., availability of perturbation tools, cellular localization of the protein) [111].
- Output: A shortlist of 4-6 high-priority candidate genes for experimental testing.
2. siRNA-Mediated Knockdown:
- Cell Culture: Maintain relevant primary cells, such as Human Umbilical Vein Endothelial Cells (HUVECs), under standard conditions.
- Transfection: For each candidate gene, transfert cells with three different non-overlapping small interfering RNAs (siRNAs) to target the gene of interest. Include appropriate negative control siRNAs (e.g., non-targeting scramble siRNA).
- Efficiency Validation: After 48-72 hours, harvest cells. Validate knockdown efficiency at the RNA level using quantitative PCR (qPCR) and, for the most effective siRNAs, at the protein level via Western Blotting [111].
3. Phenotypic Assays:
- Migration Assay: Use a wound healing ("scratch") assay. Create a uniform scratch in a confluent cell monolayer and monitor cell migration into the wound area over 12-24 hours using live-cell imaging. Compare the migration rate of knockdown cells to control cells [111].
- Sprouting Assay: Perform a 3D fibrin gel bead assay. Seed siRNA-treated endothelial cells on microcarrier beads, embed them in a fibrin gel, and stimulate sprouting with pro-angiogenic factors. After several days, quantify the number and length of sprouts emanating from each bead to assess vessel formation capacity [111].
- Proliferation Assay: Measure cell proliferation using 3H-Thymidine incorporation or other DNA synthesis-based assays (e.g., EdU staining) to determine if gene knockdown impacts cell division [111].
4. Data Analysis:
- Quantification: Use image analysis software to quantify migration and sprouting parameters.
- Statistics: Compare the phenotypic outcomes of the gene-specific siRNA groups to the negative control group using appropriate statistical tests (e.g., t-test, ANOVA). A significant impairment in migration or sprouting confirms the gene's functional role in the tested biological process.
Diagram 1: Functional validation workflow for candidate genes from scRNA-seq data.
The Scientist's Toolkit: Key Reagent Solutions
Successful functional validation relies on a suite of reliable research reagents. The following table details essential materials and their applications in the featured experimental protocols.
Table 3: Key research reagents for functional validation experiments
| Research Reagent | Function in Validation Experiments | Example Application in Protocol |
|---|---|---|
| Small Interfering RNAs (siRNAs) | Sequence-specific knockdown of target gene expression by mediating RNA interference [111] | Transfection into HUVECs to reduce expression of candidate genes like CD93 and ADGRL4 [111] |
| Primary Cells (e.g., HUVECs) | Biologically relevant in vitro model systems that retain key characteristics of their tissue of origin [111] | Used as the cellular model for siRNA knockdown and subsequent phenotypic assays in angiogenesis research [111] |
| qPCR Assays | Quantitatively measure the level of target gene mRNA expression to confirm knockdown efficiency [111] | Validation of reduced candidate gene expression post-siRNA transfection [111] |
| Fibrin Gel Matrix | A 3D extracellular matrix that supports the formation of endothelial cell sprouts, mimicking in vivo conditions [111] | Used in the 3D sprouting assay to quantify the vessel-forming capacity of endothelial cells after gene knockdown [111] |
| 3H-Thymidine / EdU | Radioactive or click-chemistry-based labels that are incorporated into newly synthesized DNA, serving as a proxy for cell proliferation [111] | Used to assess the impact of gene knockdown on endothelial cell proliferation rates [111] |
Building a Reproducible Bridge Across the "Valley of Death"
To overcome the high failure rates in translation, a paradigm shift is needed. The drug-development pipeline must integrate structured phases for assessing replicability and functional relevance before candidates proceed to costly human trials [108]. This approach mirrors the multi-phase process of clinical research.
A robust strategy involves:
- Initial Discovery & Replication: Identifying candidate genes in a discovery cohort and replicating the statistical association in an independent, similar population [109].
- Independent Laboratory Replication: Requiring successful replication of key findings by an independent laboratory, a model used by the NIH Somatic Cell Genome Editing (SCGE) Consortium [108].
- Functional Validation: Conducting targeted experiments, as detailed in this guide, to establish biological mechanism and causality [111].
- Assessment in Multiple Models: Testing the findings in other valid preclinical models to assess generalizability and limitations, as demonstrated by the Stroke Pre-Clinical Assessment Network (SPAN) [108].
This phased, cumulative approach creates a "reproducible bridge" that increases heterogeneity and confidence in a candidate gene's role, helping to ensure that only the most promising targets advance into clinical trials [108].
Diagram 2: A phased framework for bridging the "valley of death" in therapeutic development.
Primary Ovarian Insufficiency (POI) is a significant cause of female infertility, characterized by the cessation of ovarian function before age 40 and affecting 1–3.7% of women under 40 [40] [41]. While a substantial genetic basis exists for POI, with approximately 20–29% of cases having an identified genetic cause, the condition remains clinically and genetically highly heterogeneous [40] [41]. This heterogeneity presents a major challenge in moving from initial genetic associations to a mechanistic understanding of disease pathology. The research community has identified over 50 candidate genes impacting diverse biological processes including gonadal development, DNA replication/meiosis, DNA repair, transcription, signal transduction, RNA metabolism, and mitochondrial function [40] [41]. However, the functional validation of these candidates requires a multi-layered approach that integrates genomic, transcriptomic, and proteomic evidence to establish true biological causality and identify promising therapeutic targets.
Genetic Association Studies: Discovery and Limitations
Established Genetic Associations in POI
Genetic association studies have successfully identified numerous POI-linked genes and chromosomal regions. These discoveries come from various approaches, including genome-wide association studies (GWAS), rare-variant burden tests, and studies of chromosomal abnormalities.
Table 1: Key Genetic Associations in Primary Ovarian Insufficiency
| Gene/Region | Association Type | Functional Role | Evidence Level |
|---|---|---|---|
| X Chromosome (POF1, POF2, POF3 regions) | Chromosomal rearrangement/CNV [6] | Ovarian function, X-inactivation [6] | Strong (Critical regions identified) |
| FMR1 (Fragile X) | Trinucleotide repeat expansion [6] | RNA metabolism, premature follicular depletion [6] | Established (Current screening standard) |
| Turner Syndrome (45,X) | Aneuploidy [6] [40] | Gene dosage, oocyte loss [6] | Strong (4-5% of POI cases) |
| Autoimmune Regulator (AIRE) | Gene mutation (APS-1 syndrome) [40] | Immune tolerance, lymphocytic oophoritis [40] | Strong (~41% of APS-1 patients) |
| Galactose-1-Phosphate Uridylyltransferase (GALT) | Gene mutation (Galactosemia) [40] | Galactose metabolism, follicular atresia [40] | Strong (80-90% of patients) |
| BRCA2, FANCM, HELQ, ERCC6 | Gene mutation (Isolated or syndromic) [41] | DNA damage repair, meiotic integrity [41] | Emerging (Cohort evidence) |
| BNC1, BMPR1A/B, ESR2 | Gene mutation [41] | Follicular development, signaling [41] | Emerging (Cohort evidence) |
Methodological Considerations in Gene Discovery
The two primary approaches for gene discovery—GWAS and rare-variant burden tests—systematically rank genes differently, each highlighting distinct aspects of trait biology [32]. Understanding these methodological differences is crucial for interpreting candidate gene lists. GWAS typically identifies common variants with small effect sizes, while rare-variant tests pinpoint genes with larger effect sizes but lower population frequency. A recent analysis suggests that specificity, length, and random biological factors all contribute to how these studies rank genes, indicating that both methods are complementary rather than contradictory [32]. Furthermore, the candidate gene approach, which tests specific biological hypotheses, continues to yield important discoveries, particularly when guided by insights from animal models or linkage analyses [112].
Bridging the Gap: From Genetic Variant to RNA and Protein Expression
The RNA-Protein Correlation Challenge
A critical step in validating POI candidate genes involves demonstrating functional consequences at the protein level. However, the relationship between RNA transcript levels and functional protein abundance is complex and gene-specific.
Research has established that transcript and protein levels do not correlate well unless a gene-specific RNA-to-protein (RTP) conversion factor is introduced [113]. This RTP ratio varies dramatically—from a few hundred protein copies per mRNA molecule for some genes to several hundred thousand for others [113]. This variability means that mRNA levels alone are insufficient proxies for protein activity in the context of POI pathogenesis.
Chromatin Accessibility as a Regulatory Bridge
Multi-omics approaches integrating chromatin accessibility, RNA abundance, and protein levels provide a powerful framework for understanding regulatory mechanisms. Studies in human cancer models have revealed that local chromatin structure, particularly enhancers located within gene-bodies, highly correlates with coordinated RNA and protein expression [114]. This relationship is independent of overall transcriptional activity, suggesting that chromatin accessibility data can help predict which genetic variants likely result in functional protein-level consequences.
Table 2: Multi-Omics Integration Methods for POI Candidate Validation
| Methodology | Application | Key Insight for POI Research | Reference |
|---|---|---|---|
| Targeted Proteomics (PRM) | Absolute protein quantification [113] | Enables precise measurement of protein copy numbers per cell from candidate genes | [113] |
| ATAC-seq | Chromatin accessibility profiling [114] | Identifies regulatory elements predictive of correlated RNA-protein expression | [114] |
| RNA-Protein Correlation Analysis | Multi-omics integration [114] | Differentially expressed genes show higher RNA-protein correlation (median r=0.36) | [114] |
| RNA-Binding Protein (RBP) Atlas | RNA-protein interaction mapping [115] | Identifies non-classical RNA-binding domains in metabolic proteins | [115] |
Figure 1: Multi-Omics Validation Workflow for POI Candidate Genes. This framework integrates evidence from genomic variation through functional protein consequences, highlighting both canonical (solid arrows) and alternative (dashed arrows) pathways.
Experimental Protocols for Functional Validation
Targeted Proteomics for Absolute Protein Quantification
Objective: To precisely measure absolute protein copy numbers per cell for POI candidate genes across relevant tissue models.
Methodology:
- Sample Preparation: Generate cell lysates from ovarian granulosa cell models, primary oocytes, or other relevant tissue sources.
- Standard Spike-in: Use stable isotope-labeled recombinant protein fragments (QPrESTs) as internal standards, spiked at concentrations approximating a 1:1 ratio with endogenous target proteins [113].
- Digestion and Analysis: Perform trypsin digestion followed by Parallel Reaction Monitoring (PRM) mass spectrometry analysis [113].
- Cell Number Normalization: Implement histone-based normalization (H2A, H2B, H3, H4) using PRM to establish absolute cell counts in complex tissue samples [113].
- Calculation: Determine absolute protein copy numbers per cell from the ratio between sample peptides and standard peptides.
Application to POI: This protocol enables researchers to determine whether genetic variants in candidate genes result in altered protein abundance, providing direct functional evidence for pathogenicity.
Chromatin Accessibility- RNA-Protein Integration Analysis
Objective: To identify whether POI genetic variants disrupt regulatory elements that coordinate RNA and protein expression.
Methodology:
- Multi-omics Profiling: Perform simultaneous ATAC-seq (chromatin accessibility), RNA-seq (transcript abundance), and shotgun proteomics (protein abundance) on patient-matched normal and disease-relevant tissues [114].
- Regulatory Element Mapping: Categorize chromatin accessibility peaks as promoter regulatory elements (-1000 to +100 bp from TSS) or non-promoter regulatory elements [114].
- Peak-Gene Linkage: Assign regulatory elements to gene targets using correlation-based algorithms between ATAC-seq peaks and RNA expression [114].
- Correlation Analysis: Calculate sample-wise correlations between: a) chromatin accessibility and RNA abundance, and b) RNA abundance and protein abundance [114].
- Identification of Coordinated Regulation: Focus on gene-regulatory element pairs with high paired correlation values (>0.5) in both relationships, indicating coordinated regulation across multiple molecular layers.
Application to POI: This approach helps determine whether non-coding variants in POI candidates likely disrupt coordinated gene expression or represent silent polymorphisms.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for POI Candidate Gene Validation
| Reagent / Solution | Function | Application in POI Research |
|---|---|---|
| Stable Isotope-Labeled QPrESTs | Internal standards for absolute protein quantification [113] | Precisely measure protein copy numbers from POI candidate genes |
| SID-1 Similar Reagents | Facilitate dsRNA uptake in model systems [116] | Study RNA-based mechanisms and intergenerational inheritance in POI |
| Histone H2A, H2B, H3, H4 Antibodies | Cell number normalization in complex tissues [113] | Standardize protein measurements in ovarian tissue samples |
| CRISPR Interference (CRISPRi) Screens | Systematic knockdown of gene networks [117] | Identify regulators of androgen receptor and other pathways relevant to POI |
| Parallel Reaction Monitoring (PRM) Assays | Targeted protein quantification [113] | Validate protein-level effects of genetic variants in POI candidates |
| Spatially Resolved Multi-omics Platforms | Simultaneous transcriptomic, metabolomic, proteomic analysis [117] | Characterize role of specific ovarian cell types (e.g., lipid-associated macrophages) |
Signaling Pathways and Intergenerational Regulation
Expanding the Paradigm: RNA-Based Inheritance
Recent research has revealed unexpected mechanisms of gene regulation with potential significance for POI. Studies in C. elegans have identified that double-stranded RNA (dsRNA) can travel between cells and across generations, with the SID-1 protein acting as a key regulator of this process [116]. Interestingly, removal of SID-1 resulted in more persistent heritable changes in gene expression across over 100 generations, suggesting a complex relationship between RNA transport and epigenetic stability [116]. Additionally, the discovery of sdg-1, a gene located within a "jumping gene" that helps regulate the movement of these genetic elements, reveals another layer of potential genomic instability relevant to ovarian function [116].
Figure 2: TGFβ Immunosuppression Pathway in Cancer Metastasis with Parallels to POI. This pathway, discovered in colorectal cancer, demonstrates how a single signaling molecule (TGFβ) can suppress immune response through two parallel mechanisms—similar to potential multi-mechanism actions in POI pathogenesis [117].
The integration of genetic association data with functional evidence at the RNA and protein levels represents the most promising path forward for unraveling POI pathophysiology. Large cohort studies have demonstrated the high yield of comprehensive genetic testing, with pathogenic findings in 29.3% of POI cases [41]. Beyond diagnosing the cause of infertility, genetic characterization enables personalized medicine approaches to prevent comorbidities (particularly important for the 37.4% of cases with tumor/cancer susceptibility genes) and predict residual ovarian reserve (relevant for 60.5% of cases) [41]. Furthermore, the identification of new pathways including NF-κB signaling, post-translational regulation, and mitophagy provides novel therapeutic targets for future intervention [41]. As RNA-based therapeutics advance, understanding the natural mechanisms of RNA transport and regulation will be crucial for developing effective treatments for this complex and impactful condition.
Conclusion
Successful replication of candidate gene studies is a cornerstone of credible genetic research but remains inherently challenging. This synthesis underscores that rigorous methodology, adequate power, and transparent reporting are paramount. The future of the field lies not in abandoning candidate genes, but in integrating them into more complex models of gene-environment interplay and polygenic effects. For drug development, conclusively replicated genes with large effect sizes offer valuable targets, but their translation requires a multi-level validation pipeline from genetic association to cellular function. Embracing collaborative, well-powered, and pre-registered studies will be essential for building a cumulative and trustworthy body of knowledge that reliably informs biomedical science and therapeutic development.
References
- 1. How can I be sure a genetic test is valid and useful? [https://medlineplus.gov/genetics/understanding/testing/validtest/]
- 2. Genetic Test Assessment - An Evidence Framework for ... - NCBI [https://www.ncbi.nlm.nih.gov/books/NBK425803/]
- 3. Regulation of Genetic Tests [https://www.genome.gov/about-genomics/policy-issues/Regulation-of-Genetic-Tests]
- 4. Landscape of pathogenic mutations in premature ovarian ... [https://www.nature.com/articles/s41591-022-02194-3]
- 5. Human Genetics Research Presentations and Resources [https://www.thermofisher.com/us/en/home/life-science/sequencing/sequencing-applications/complex-disease/human-genetics-research/human-genetics-presentations-resources.html]
- 6. investigating the genetics of primary ovarian insufficiency [https://pmc.ncbi.nlm.nih.gov/articles/PMC11603650/]
- 7. Reactions: Solving the replication crisis - C&EN [https://cen.acs.org/research-integrity/reproducibility/Reactions-Solving-replication-crisis/103/web/2025/07]
- 8. A Re-evaluation of Candidate Gene Studies for Well-Being ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9356956/]
- 9. Candidate Gene Studies of a Promising Intermediate ... [https://www.nature.com/articles/npp2012245]
- 10. Lessons from complex trait genetics may help us overcome ... [https://www.sciencedirect.com/science/article/pii/S0010945223001971]
- 11. Giant study finds a research field that's mostly reproducible [https://www.nature.com/articles/d41586-025-02250-1]
- 12. Replication Study: Discovery and preclinical validation of ... [https://elifesciences.org/articles/17044]
- 13. Replicating scientific results is tough — but essential [https://www.nature.com/articles/d41586-021-03736-4]
- 14. Research replication can determine how well science is ... [https://theconversation.com/research-replication-can-determine-how-well-science-is-working-but-how-do-scientists-replicate-studies-260771]
- 15. Feasibility of Using Real-World Data to Replicate Clinical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6802419/]
- 16. How Psychological Study Results Have Changed Since ... [https://www.psychologicalscience.org/publications/observer/results-changed-replication-crisis.html]
- 17. Publication bias and the canonization of false facts - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5173326/]
- 18. Publication Biases in Replication Studies [https://papers.ssrn.com/sol3/papers.cfm?abstract_id=3454901]
- 19. How to interpret the results of replication studies [https://replicationindex.com/2018/02/08/replicability-101-how-to-interpret-the-results-of-replication-studies/]
- 20. Why is Replication in Research Important? [https://www.aje.com/arc/why-is-replication-in-research-important]
- 21. Validating Genome-Wide Association Candidates Controlling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5291020/]
- 22. How Much Should We Spend on Scientific Replication? [https://ifp.org/how-much-should-we-spend-on-replication/]
- 23. Publication bias and the failure of replication in ... [https://pubmed.ncbi.nlm.nih.gov/23055145/]
- 24. From Candidate Genes to Genome-wide Association [https://pmc.ncbi.nlm.nih.gov/articles/PMC4617623/]
- 25. How genome-wide association studies (GWAS) made ... [https://www.nature.com/articles/s41386-019-0389-5]
- 26. Candidate genes versus genome-wide associations [https://pmc.ncbi.nlm.nih.gov/articles/PMC3049081/]
- 27. Genome-wide association study of childhood B-cell acute ... [https://www.nature.com/articles/s41467-025-64337-7]
- 28. Genome-Wide Association Study and Meta-Analysis Uncovers ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12385673/]
- 29. Genome-wide association studies with experimental ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11282472/]
- 30. Editing GWAS: experimental approaches to dissect and ... [https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-021-00857-3]
- 31. A replication study of novel fetal hemoglobin-associated ... [https://pubmed.ncbi.nlm.nih.gov/39886999]
- 32. How do genetic association studies rank genes? [https://www.nature.com/articles/d41586-025-03651-y]
- 33. Exploring dopamine as the master regulator of brain circuitry ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12311831/]
- 34. Decoding serotonin: the molecular symphony behind ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12058683/]
- 35. Dopamine and Serotonin Transporter Genes Regulation in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11429336/]
- 36. Dopaminergic and serotonergic genetic variants predict ... [https://www.nature.com/articles/s41598-025-03772-4]
- 37. Effect of individual variations in genes related to dopamine ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0314173]
- 38. Brain-Region-Specific Genes Form the Major Pathways ... [https://www.mdpi.com/1422-0067/25/5/2882]
- 39. Serotonin modulation of metabolism and stress response in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12577022/]
- 40. Genetic insights into the complexity of premature ovarian ... [https://rbej.biomedcentral.com/articles/10.1186/s12958-024-01254-2]
- 41. Genetic landscape of a large cohort of Primary Ovarian ... [https://www.sciencedirect.com/science/article/pii/S2352396422004285]
- 42. Genetics of primary ovarian insufficiency: new developments ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4594617/]
- 43. Overcoming the Winner's Curse: Estimating Penetrance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1852705/]
- 44. Sample size, power and effect size revisited: simplified and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7745163/]
- 45. Genetics Investigation of Idiopathic Premature Ovarian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11942377/]
- 46. Exploring biomarkers of premature ovarian insufficiency ... [https://www.nature.com/articles/s41598-023-38754-x]
- 47. Immune-molecular nexus in reproductive disorders [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1652519/full]
- 48. Genetic landscape of a large cohort of Primary Ovarian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9475279/]
- 49. Identification of new variants and candidate genes in women ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9723088/]
- 50. Environment-to-phenotype mapping and adaptation strategies ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6628789/]
- 51. mechanisms linking POI and RSA - PMC - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC12515496/]
- 52. Reproducibility, replication, and preregistration [https://sciety-discovery.elifesciences.org/articles/by?article_doi=10.31234/osf.io/drvnz_v1]
- 53. The effect of preregistration on trust in empirical research ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7211853/]
- 54. The preregistration revolution - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5856500/]
- 55. Preregistration [https://www.cos.io/initiatives/prereg]
- 56. Choosing the Right Preregistration Template: A Guide for Researchers [https://www.cos.io/blog/choosing-preregistration-template-guide-for-researchers]
- 57. and Registered Reports | University of Surrey Preregistration [https://www.surrey.ac.uk/library/open-research/preregistration-and-registered-reports]
- 58. Comparison of Preregistration Platforms - Inria - Institut national de recherche en sciences et technologies du numérique [https://inria.hal.science/hal-03588379]
- 59. Comparison of Preregistration Platforms | FORRT - Framework for Open and Reproducible Research Training [https://forrt.org/curated_resources/comparison-of-preregistration-platforms/]
- 60. Benefits and Concerns of Registration - OSF Support [https://help.osf.io/article/603-benefits-of-registration]
- 61. Preregistration and reproducibility [https://www.sciencedirect.com/science/article/pii/S0167487018302903]
- 62. The ethics of collaborative authorship - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC3166465/]
- 63. A conceptual framework for implementation fidelity [https://implementationscience.biomedcentral.com/articles/10.1186/1748-5908-2-40]
- 64. Genetic factors behind impulsive choices tied to metabolic ... [https://www.news-medical.net/news/20251124/Genetic-factors-behind-impulsive-choices-tied-to-metabolic-and-mental-health.aspx]
- 65. The brief resilience scale: a genome-wide association ... [https://pubmed.ncbi.nlm.nih.gov/41013467/]
- 66. Causal and Candidate Gene Variants in a Large Cohort ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9006976/]
- 67. Contextual sensitivity in scientific reproducibility - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4988618/]
- 68. Why the Journal of Personality and Social Psychology ... [https://replicationindex.com/2018/01/05/bem-retraction/]
- 69. Addressing replicability concerns via adversarial ... [https://www.sciencedirect.com/science/article/abs/pii/S0022103117306352]
- 70. Clustering of lymphoid neoplasms by cell of origin, somatic ... [https://www.nature.com/articles/s41408-025-01351-4]
- 71. Gene–environment interactions and their impact on human ... [https://www.nature.com/articles/s41435-022-00192-6]
- 72. Testing different types of genotype-environment correlation [https://pmc.ncbi.nlm.nih.gov/articles/PMC3093315/]
- 73. Candidate gene-environment interactions in substance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10617739/]
- 74. Will genomics revolutionise research on gene–environment ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9804537/]
- 75. An approach to identify gene-environment interactions and ... [https://www.nature.com/articles/s41467-024-47806-3]
- 76. Gene-environment interplay in early life cognitive ... [https://www.sciencedirect.com/science/article/pii/S0160289623000296]
- 77. An expanded view of complex traits: from polygenic to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5536862/]
- 78. Polygenic Risk Scores [https://www.genome.gov/Health/Genomics-and-Medicine/Polygenic-risk-scores]
- 79. Monogenic, Oligogenic, and Polygenic... what's the ... [https://www.genomicmd.com/blog/monogenic-oligogenic-and-polygenic...-whats-the-difference]
- 80. Polygenic architecture of rare coding variation across 394,783 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10614218/]
- 81. Statistical selection of biological models for genome-wide ... [https://www.sciencedirect.com/science/article/abs/pii/S1046202317304966]
- 82. A guide to performing Polygenic Risk Score analyses - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7612115/]
- 83. Performance of deep-learning-based approaches to ... [https://www.nature.com/articles/s41467-025-60056-1]
- 84. A dynamic model for genome-wide association studies - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3103104/]
- 85. Replication problems are not competence problems [https://thenewstatistics.com/itns/2017/05/01/replication-problems-are-not-competence-problems/]
- 86. Incomplete Coverage of Candidate Genes: A Poorly ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2647155/]
- 87. Commentary: Fundamental problems with candidate gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5312579/]
- 88. Ten simple rules for designing and conducting undergraduate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10019630/]
- 89. Integrative genomic and single-cell framework identifies ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12149120/]
- 90. Comprehensive interrogation of synthetic lethality in the ... [https://www.nature.com/articles/s41586-025-08815-4]
- 91. Single cell multiomics approach to analyze replication ... [https://www.nature.com/articles/s42003-025-08694-5]
- 92. Quantitative Data Analysis. A Complete Guide [2025] [https://www.6sigma.us/six-sigma-in-focus/quantitative-data-analysis/]
- 93. Aberrant downregulation of Y-box binding protein 1 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12095738/]
- 94. Current status and future prospects of mesenchymal stem cell ... [https://ovarianresearch.biomedcentral.com/articles/10.1186/s13048-025-01813-0]
- 95. Genome-wide association study and fine-mapping using ... [https://www.sciencedirect.com/science/article/pii/S0022030225007246]
- 96. Genome-wide association studies and candidate genes ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11744-1]
- 97. A meta-analysis of genome-wide association studies ... [https://www.nature.com/articles/s41598-025-00819-4]
- 98. Genome-Wide Association Study Revealed Candidate Genes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12561659/]
- 99. Genome-wide analysis of genetic loci and candidate genes ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1593395/full]
- 100. Genome and transcriptome wide association study identify ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1606220/full]
- 101. AGT, CYP11B2 & ADRB2 gene polymorphism & essential ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11463865/]
- 102. exploring the role of genetics, epigenetics, microbiome, and ... [https://jhpn.biomedcentral.com/articles/10.1186/s41043-025-01058-z]
- 103. 醛固酮合成酶(CYP11B2)基因T(-344)C多态性与高血压病相关 ... [http://zhgxyzz.xml-journal.net/article/doi/10.16439/j.cnki.1673-7245.2003.03.014]
- 104. Effect of ACE, ACE2 and CYP11B2 gene polymorphisms and ... [https://bmcmedgenomics.biomedcentral.com/articles/10.1186/s12920-022-01177-0]
- 105. Contribution of CYP11B2, REN and AGT genes in genetic ... [https://pure.johnshopkins.edu/en/publications/contribution-of-cyp11b2-ren-and-agt-genes-in-genetic-predispositi-3]
- 106. Discovery and in Vivo Evaluation of Potent Dual CYP11B2 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4027133/]
- 107. 肾素-血管紧张素-醛固酮系统基因多态性与老年高血压患者 ... [https://www.ebiotrade.com/newsf/2025-8/20250821002952210.htm]
- 108. Building reproducible bridges to cross the “valley of death” [https://www.jci.org/articles/view/177383]
- 109. Replication and Validation in the 'Omics Era - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC7408356/]
- 110. Functional Validation of Candidate Genes Detected by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5940157/]
- 111. Prioritization and functional validation of target genes from ... [https://www.nature.com/articles/s42003-023-05006-7]
- 112. The Candidate Gene Approach - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6709736/]
- 113. Gene‐specific correlation of RNA and protein levels in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5081484/]
- 114. Chromatin accessibility associates with protein-RNA ... [https://www.nature.com/articles/s41467-021-25872-1]
- 115. Uncovering the complexity of RNA–protein interactions [https://www.nature.com/articles/s41580-025-00859-8]
- 116. New Study Reveals How RNA Travels Between Cells to ... [https://cmns.umd.edu/news-events/news/new-study-reveals-how-rna-travels-between-cells-control-genes-across-generations]
- 117. Articles in 2025 | Nature Genetics [https://www.nature.com/ng/articles?year=2025]