Silencing Fertility: RNAi Strategies to Reduce Fecundity and Egg Hatchability in Pest and Disease Vectors
This article explores the application of RNA interference (RNAi) as a targeted strategy to suppress reproductive success by reducing fecundity and egg hatchability.
Silencing Fertility: RNAi Strategies to Reduce Fecundity and Egg Hatchability in Pest and Disease Vectors
Abstract
This article explores the application of RNA interference (RNAi) as a targeted strategy to suppress reproductive success by reducing fecundity and egg hatchability. It provides a comprehensive analysis for researchers and drug development professionals, covering the foundational science of key genetic targets, advanced methodological approaches for dsRNA delivery, solutions for common troubleshooting and optimization challenges, and rigorous validation through comparative analysis with other techniques. The review synthesizes recent advances and future directions, highlighting the transformative potential of this approach in developing novel biocontrol agents and therapeutic interventions.
The Genetic Blueprint: Identifying Key Molecular Targets for Reproductive Disruption
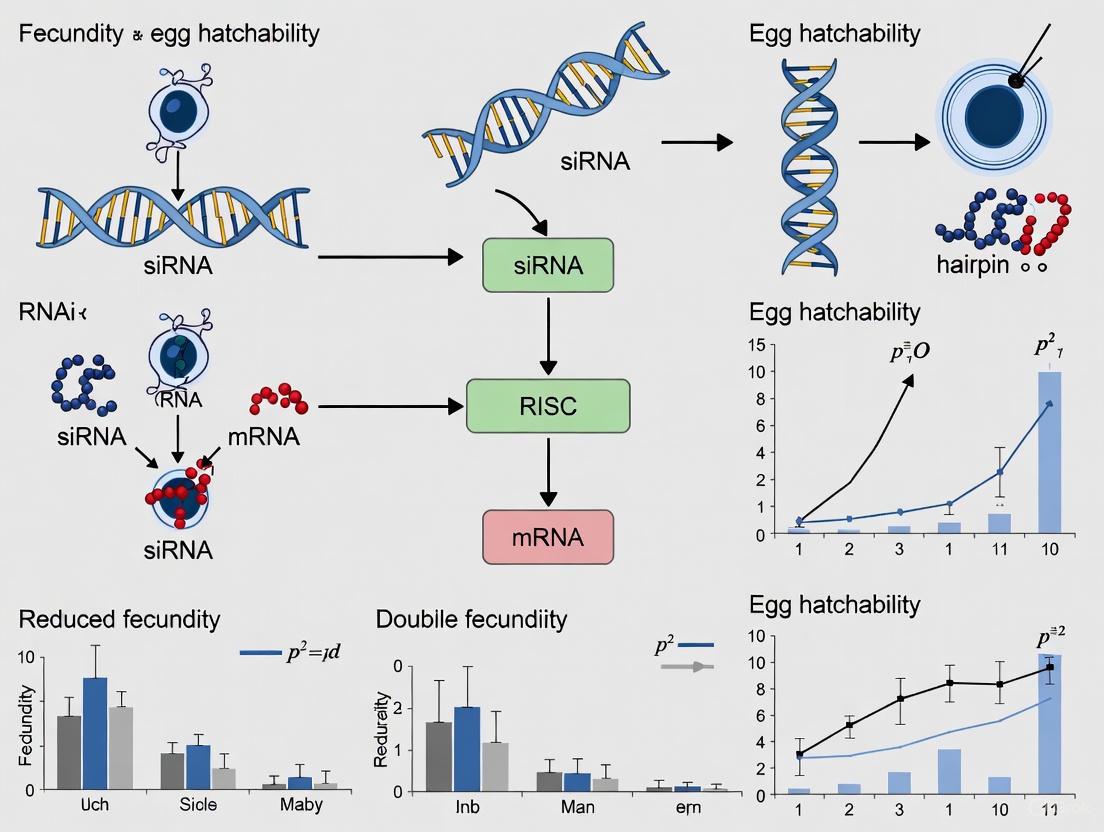
The use of RNA interference (RNAi) to reduce fecundity and egg hatchability represents a promising frontier in pest control and developmental biology research. By targeting genes essential for embryonic development, this approach enables precise suppression of pest populations at their earliest life stages, potentially before crop damage occurs. The Sl102 gene in Spodoptera littoralis (cotton leafworm) exemplifies this strategy, as its silencing disrupts embryonic development and dramatically reduces egg viability [1]. This application note details protocols for identifying and targeting such essential embryonic genes, providing researchers with methodologies to exploit RNAi for fecundity reduction in insect pests and disease vectors.
Key Experimental Data and Findings
Table 1: Quantitative Data from RNAi-Mediated Suppression of Embryogenesis
| Target Gene | Organism | RNAi Delivery Method | Effect on Embryo Hatching | Additional Fitness Impacts | Reference |
|---|---|---|---|---|---|
| Sl102 | Spodoptera littoralis | Egg soaking in dsRNA solution (250 ng/µL, 120 min) | Drastic reduction in hatching rate | High mortality of hatched larvae; significant developmental delay | [1] |
| NlATG3 | Nilaparvata lugens (Brown Planthopper) | Injection of dsRNA into nymphs (62.5-250 ng/insect) | Hatchability reduced from 95.7% to 0% in specific crosses | 80.4% reduction in total eggs laid per female; prevented molting | [2] |
| Core RNAi Machinery Genes (e.g., dcr-2, ago-2) | Diabrotica virgifera virgifera (Western Corn Rootworm) | Oral delivery via dsRNA-treated diet | Not specified | Decreased pupation ability; reduced adult emergence; diminished reproductive capacity | [3] |
Table 2: Temporal Efficacy of RNAi Targeting in Embryos
| Developmental Stage Targeted | Gene | Optimal dsRNA Concentration | Exposure Duration | Key Phenotypic Outcomes |
|---|---|---|---|---|
| Early Embryo (Egg) | Sl102 | 250 ng/µL | 120 minutes | Disrupted embryonic development, morphological alterations, reduced hatching |
| Late Larval (pre-pupation) | NlATG3 | 62.5-250 ng/insect | Single injection | Cuticle defects, loose and curved new cuticle, blocked molting |
| Multiple larval instars | dcr-1, ago-1 | Diet-mediated delivery | 2-7 days | Changes in miRNA expression, fitness costs manifesting at pupal/adult stages |
Experimental Protocols
RNAi via Egg Soaking for Embryonic Gene Silencing
This protocol, adapted from successful Sl102 silencing in Spodoptera littoralis, is suitable for lepidopteran and other insect eggs with permeable chorions [1].
Materials Required:
- Highly synchronized, newly-laid eggs (within 30-minute oviposition window)
- dsRNA targeting gene of interest (e.g., Sl102) and control dsRNA (e.g., GFP)
- Phosphate Buffered Saline (PBS 1×; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4; pH 7.4)
- 1.5 mL Eppendorf tubes
- Environmental chamber (25°C ± 1°C)
Procedure:
- Egg Collection: Collect 120 highly synchronized eggs from the same egg mass using a fine brush.
- dsRNA Solution Preparation: Prepare soaking solution with 250 ng/µL dsRNA in PBS.
- Soaking Process: Transfer eggs to 1.5 mL tube containing 50 µL dsRNA solution.
- Incubation: Soak eggs for 120 minutes at 25°C ± 1°C.
- Post-treatment Handling: Remove dsRNA solution and transfer eggs to standard diet.
- Phenotypic Assessment:
- Monitor hatching rates daily
- Assess larval mortality and developmental abnormalities
- Conduct qRT-PCR to verify target gene knockdown
dsRNA Microinjection for Nymphal/Adult Gene Silencing
This method, based on NlATG3 silencing in brown planthopper, is effective for systemic RNAi response [2].
Materials Required:
- Experimental insects (nymphs or adults)
- Purified dsRNA (62.5-250 ng/µL)
- Microinjection system (capillary puller, micromanipulator, microinjector)
- CO2 or ice anesthesia setup
Procedure:
- dsRNA Preparation: Synthesize and purify dsRNA targeting gene of interest.
- Insect Preparation: Anesthetize insects using CO2 or ice immersion.
- Microinjection: Inject 50 nL dsRNA solution (62.5-250 ng/µL) into thoracic cavity or abdomen.
- Post-injection Recovery: Transfer insects to fresh diet and monitor recovery.
- Phenotypic Assessment:
- Record mortality daily
- Assess molting defects for nymphs
- For adults: monitor fecundity (eggs laid), egg hatchability, and ovarian development
Signaling Pathways and Experimental Workflows
RNAi Experimental Workflow for Embryonic Gene Silencing
Biological Function of Sl102 and RNAi Intervention Points
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for RNAi Embryogenesis Studies
| Reagent/Material | Specifications | Application | Protocol-Specific Notes |
|---|---|---|---|
| dsRNA | HPLC-purified, 200-500 bp target-specific fragments | Gene silencing | Critical to verify sequence specificity and absence of off-target effects |
| Egg Soaking Buffer | 1× PBS, pH 7.4 | Egg permeabilization and dsRNA delivery | Optimal concentration: 250 ng/µL; exposure: 120 minutes [1] |
| Microinjection System | Capillary puller, micromanipulator, nanoliter injector | Precise dsRNA delivery into insects | Injection volume: ~50 nL; dsRNA concentration: 62.5-250 ng/µL [2] |
| qRT-PCR Reagents | SYBR Green master mix, gene-specific primers | Knockdown validation | Primers should flank dsRNA target region; include reference genes |
| Artificial Diet | Species-specific formulation | Post-treatment insect maintenance | Critical for assessing fitness costs and transgenerational effects |
Targeting essential embryonic genes like Sl102 through RNAi represents a highly specific approach for reducing fecundity and egg hatchability in pest insects. The protocols outlined here provide researchers with robust methodologies for implementing this strategy across various insect systems. The consistent observation that silencing key developmental genes causes not only reduced hatching but also significant fitness costs in surviving individuals [1] [2] [3] reinforces the potential of this approach for sustainable pest management solutions. Future research directions should focus on identifying additional essential embryonic genes across species, optimizing delivery methods for field applications, and investigating potential resistance mechanisms to maintain the long-term efficacy of RNAi-based control strategies.
Dopamine, a critical catecholamine neurotransmitter, plays a vital role in the neuroendocrine system, regulating essential physiological processes including reproduction. The synthesis of dopamine from the amino acid tyrosine is a two-step enzymatic process: the rate-limiting conversion of tyrosine to L-DOPA by tyrosine hydroxylase (TH), followed by the decarboxylation of L-DOPA to dopamine by the enzyme L-DOPA decarboxylase (DDC) [4]. This pathway is crucial for maintaining pregnancy and ensuring healthy embryonic development. Research indicates that downregulation of DDC and the dopamine D2 receptor (D2R) in placental trophoblasts is associated with recurrent miscarriages, reflecting a reduced catecholamine signaling cascade on the fetal side [4].
The application of RNA interference (RNAi) to silence key genes in the dopamine synthesis pathway presents a promising strategy for investigating neuroendocrine regulation and developing novel control methods for pest species by reducing fecundity and egg hatchability. This approach leverages the natural mechanism of sequence-specific post-transcriptional gene silencing mediated by double-stranded RNA (dsRNA) [5]. By targeting TH and DDC genes, researchers can disrupt dopamine production, leading to impaired reproductive outcomes, as demonstrated in recent studies on insect pest species [2] [1]. This protocol outlines detailed methodologies for applying RNAi to disrupt dopamine synthesis and provides quantitative data on its effects on fecundity and embryonic development.
Background
The Dopamine Synthesis Pathway in Neuroendocrine Regulation
The dopamine synthesis pathway is integral to the neuroendocrine system, which interfaces the nervous and endocrine systems via the hypothalamus-pituitary complex [6]. Hypothalamic neuroendocrine cells secrete neurohormones that regulate pituitary function, which in turn controls peripheral endocrine glands. Dopamine itself acts as a hypothalamic neurohormone, inhibiting prolactin synthesis by the pituitary [6]. During pregnancy, dopamine plays a significant role in human placental endocrine function, with the ability to inhibit human placental lactogen (hPL) and human chorionic gonadotropin production [4]. The expression of D2R increases with gestational week, reaching a maximum at term, and alterations in this expression have been observed in pregnancy complications such as preeclampsia [4].
RNAi as a Tool for Gene Silencing
RNA interference is a powerful reverse genetics tool for specifically silencing gene function. The process involves introducing dsRNA homologous to a target gene, which leads to the degradation of complementary mRNA through the activity of the dicer enzyme and the RNA-induced silencing complex (RISC) [5]. While RNAi efficiency can be variable, with approximately 18.5% of experiments showing insufficient silencing (fold change >0.7) [7], careful experimental design can overcome these limitations. Targeting early developmental stages such as embryos has proven particularly effective, as these stages present a less harsh degradation environment for dsRNAs [1].
Application Notes: RNAi-Mediated Suppression of Dopamine Synthesis
Experimental Rationale and Target Validation
Silencing TH and DDC genes disrupts the dopamine synthesis pathway, which can impair reproductive functions and embryonic development. Previous research has demonstrated that downregulation of DDC in placental trophoblasts is associated with recurrent miscarriages [4]. In insect models, RNAi-mediated suppression of key embryonic genes has resulted dramatically reduced egg hatch rates [2] [1].
Target validation should include:
- Expression Profiling: Confirm target gene expression during embryonic stages via qRT-PCR.
- In Silico Design: Use algorithms to design siRNA probes with higher predicted efficacy, avoiding regions with high secondary structure.
- Efficiency Screening: Implement a high-throughput screening system using reporter fusions to identify the most effective siRNA probes before proceeding with functional studies [5].
Key Parameters for Successful RNAi in Embryonic Stages
Research indicates that targeting embryonic stages requires optimization of several parameters:
- dsRNA Concentration: Empirical testing of concentrations (typically 50-250 ng/μL) is necessary [1].
- Delivery Timing: Soaking experiments should be timed to coincide with peak expression of target genes during embryogenesis [1].
- Soaking Duration: Treatment duration (30-120 minutes) significantly impacts silencing efficiency and embryo survival [1].
Table 1: Quantitative Effects of RNAi on Embryonic Survival and Hatch Rates
| Target Gene | Organism | dsRNA Concentration | Soaking Duration | Hatch Rate Reduction | Additional Effects |
|---|---|---|---|---|---|
| Sl102 (Amyloid fibril gene) | Spodoptera littoralis | 250 ng/μL | 120 min | Drastic reduction | 80.4% reduction in eggs laid per female [1] |
| NlATG3 (Autophagy-related) | Nilaparvata lugens (Brown planthopper) | 62.5-250 ng per insect | - | Hatchability reduced from 95.7% to 0% in dsNlATG3 × dsGFP [2] | 100% mortality of 5th-instar nymphs within 5 days [2] |
| DDC (Dopamine synthesis) | Human placental cells | - | - | Associated with recurrent miscarriages [4] | Downregulation in trophoblasts and decidua [4] |
Table 2: RNAi Silencing Efficiency Across Validation Methods and Cell Lines
| Validation Method | Average Fold Change | Notes |
|---|---|---|
| Western Blot | 0.43 | Highest efficiency among validation methods [7] |
| Quantitative PCR (qPCR) | 0.47 | Intermediate efficiency [7] |
| Microarray | 0.55 | Lower efficiency [7] |
| Cell Line | Average Fold Change | Notes |
| MCF7 | 0.59 | Lowest silencing efficiency [7] |
| SW480 | 0.30 | Highest silencing efficiency [7] |
Experimental Protocols
Protocol 1: dsRNA Production and Validation
Objective: To produce and validate dsRNA targeting TH and DDC genes.
Materials:
- Template DNA for target genes (TH or DDC)
- T7 or T3 RNA polymerase
- DNase I (RNase-free)
- PCR purification kit
- Nuclease-free water
Procedure:
- Template Preparation: Amplify target sequences (300-500 bp) from cDNA using gene-specific primers with appended T7 or T3 promoter sequences.
- In Vitro Transcription: Perform transcription reaction with T7 or T3 RNA polymerase according to manufacturer's instructions.
- DNase Treatment: Incubate with DNase I (15 min, 37°C) to remove template DNA.
- dsRNA Purification: Purify dsRNA using phenol:chloroform extraction or commercial purification kits.
- Quality Control: Verify dsRNA integrity by agarose gel electrophoresis and quantify using spectrophotometry.
- Validation: Test silencing efficiency in a reporter system before proceeding to functional experiments [5].
Protocol 2: Egg Soaking for Embryonic RNAi
Objective: To deliver dsRNA to embryonic stages via soaking for gene silencing.
Materials:
- Synchronized eggs (laid within 30-minute interval)
- dsRNA targeting TH or DDC (50-250 ng/μL in PBS)
- PBS (Phosphate Buffered Saline 1×; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4; pH 7.4)
- 1.5 mL Eppendorf tubes
Procedure:
- Egg Collection: Collect approximately 120 highly synchronized eggs in a 1.5 mL Eppendorf tube [1].
- Soaking Solution Preparation: Prepare dsRNA solution in PBS at desired concentration (50, 100, or 250 ng/μL) [1].
- Treatment: Add 50 μL of dsRNA solution to the eggs, ensuring complete immersion.
- Incubation: Soak eggs for 30, 60, or 120 minutes at 25°C ± 1 [1].
- Post-treatment: Remove soaking solution and transfer eggs to appropriate incubation conditions.
- Monitoring: Record egg hatching rates and observe developmental abnormalities.
- Efficiency Validation: Extract RNA from a subset of eggs at 24-48 hours post-treatment and assess target gene expression via qRT-PCR.
Protocol 3: Validation of Silencing Efficiency
Objective: To quantify the efficiency of RNAi-mediated silencing of target genes.
Materials:
- TRIzol Reagent for RNA extraction
- Retrotranscription kit
- qPCR reagents and system
- Gene-specific primers for TH, DDC, and reference genes
Procedure:
- RNA Extraction: Homogenize tissue samples in TRIzol and extract total RNA according to manufacturer's instructions [1].
- RNA Quantification: Determine concentration and purity using a spectrophotometer [1].
- cDNA Synthesis: Perform retrotranscription with 200 ng/μL total RNA [1].
- qPCR Setup: Prepare reactions with gene-specific primers and SYBR Green master mix.
- Amplification: Run qPCR with appropriate cycling conditions.
- Data Analysis: Calculate fold change using the 2^(-ΔΔCt) method relative to control groups (e.g., dsGFP-treated samples).
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for RNAi Experiments Targeting Dopamine Synthesis
| Reagent / Material | Function | Application Notes |
|---|---|---|
| T7/T3 RNA Polymerase | In vitro transcription of dsRNA | Essential for producing high-quality dsRNA probes [5] |
| DNase I (RNase-free) | Removal of template DNA after transcription | Preces false positives in downstream applications [1] |
| PBS (1×, pH 7.4) | Buffer for dsRNA delivery in soaking experiments | Maintains osmotic balance during embryonic treatments [1] |
| TRIzol Reagent | Total RNA extraction from tissues | Preserves RNA integrity for accurate expression analysis [1] |
| Retrotranscription Kit | cDNA synthesis from RNA templates | Enables gene expression analysis via qPCR [1] |
| SYBR Green Master Mix | Fluorescent detection in qPCR | Allows quantitative assessment of silencing efficiency [1] |
| dsGFP Control | Negative control for RNAi experiments | Controls for non-specific effects of dsRNA treatment [1] |
Signaling Pathways and Experimental Workflows
Diagram 1: Dopamine Synthesis Pathway and RNAi Interference Point. This diagram illustrates the enzymatic pathway for dopamine synthesis and the points where RNAi-mediated silencing disrupts the process, leading to impaired reproductive outcomes.
Diagram 2: RNAi Experimental Workflow for Embryonic Silencing. This workflow outlines the key steps for implementing RNAi-mediated silencing of dopamine synthesis genes in embryonic stages, from probe design to outcome analysis.
RNA interference (RNAi) technology has emerged as a transformative approach for sustainable pest management by enabling sequence-specific silencing of genes essential for insect development and reproduction [8]. Within this paradigm, targeting vital metabolic and structural proteins critical for embryonic development presents a particularly powerful strategy for suppressing pest populations by reducing both fecundity and egg hatchability. This application note details current protocols and mechanistic insights into disrupting key physiological processes during oogenesis and embryogenesis through RNAi-mediated gene silencing, providing researchers with practical methodologies for developing next-generation pest control solutions. By focusing on genes encoding proteins fundamental to chitin biosynthesis, hormone signaling, and structural integrity, this approach offers a precise biological tool that aligns with growing demands for reduced environmental pesticide loads and targeted species-specific control mechanisms [1] [8].
Key Targets for Disrupting Egg Development and Integrity
High-Value Target Genes and Their Phenotypic Impacts
Table 1: Key Target Genes for RNAi-Mediated Suppression of Egg Development
| Target Gene | Insect Species | Biological Function | RNAi-Induced Phenotype | Efficacy (Hatch Reduction) |
|---|---|---|---|---|
| Sl102 | Spodoptera littoralis | Encodes precursors of functional amyloid fibrils; immune response and basal lamina formation | Drastic reduction in egg hatching; high mortality of hatched larvae; developmental delays and morphological alterations [1] | Strong reduction (Peak expression 32h after oviposition) [1] |
| CYP303A1 | Nilaparvata lugens | Cytochrome P450 enzyme; regulation of hatching-related genes | Significant reduction in egg hatchability; abnormal embryonic development; delayed eyespot formation; dispersed yolk granules [9] | Significant reduction (Prolonged embryonic period) [9] |
| LmGFAT | Locusta migratoria | Rate-limiting enzyme in hexosamine pathway; chitin biosynthesis | 95% egg developmental arrest; failure of molting in nymphal stage [8] | 95% egg mortality [8] |
| LsTH/LsDDC | Laodelphax striatellus | Dopamine synthesis enzymes; regulation of vitellogenin and hormone signaling | Reduced fecundity; inhibited egg hatchability and development; downregulated Vg and JH/20E pathway genes [10] | Significant inhibition of hatching and development [10] |
Enhanced Delivery Formulations for Improved RNAi Efficacy
Table 2: Delivery Strategies for RNAi in Insect Embryos
| Delivery Method | Target Species | Formulation | Advantages | Efficacy Enhancement |
|---|---|---|---|---|
| Egg Soaking | Spodoptera littoralis | dsRNA in PBS solution (250 ng/μL) [1] | Non-invasive; bypasses egg barriers; suitable for high-throughput screening | High silencing efficiency with 120 min soaking [1] |
| Chitosan-complexed dsRNA | Locusta migratoria | Chitosan-nanoparticle formulated dsRNA [8] | Protects dsRNA from degradation; improves cellular uptake; enhances environmental stability | Increased mortality from 70% (naked dsRNA) to nearly 90% [8] |
| Microinjection | Triatoma infestans | dsRNA delivered via abdominal microinjection [11] | Precise dosage control; direct delivery to target tissues; bypasses digestive nucleases | Effective silencing in adult insects [11] |
Experimental Protocols
RNAi via Egg Soaking inSpodoptera littoralis
Principle: Soaking eggs in dsRNA solution enables passive uptake of dsRNA through the egg chorion, inducing gene silencing during critical embryonic developmental stages [1]. This protocol is optimized for the Egyptian cotton leafworm but can be adapted for other lepidopteran species.
Materials:
- dsRNA targeting gene of interest (e.g., Sl102)
- Phosphate-buffered saline (PBS 1×; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4; pH 7.4)
- Newly laid S. littoralis egg masses (<30 minutes old)
- 1.5 mL Eppendorf tubes
- Fine-tip brushes for egg handling
- Incubator maintained at 25°C ± 1°C
Procedure:
- Egg Collection: Collect newly laid S. littoralis eggs (within 30-minute intervals) using a fine-tip brush. Select 120 highly synchronized eggs from the same egg mass for experimental consistency [1].
- dsRNA Preparation: Dilute synthesized dsRNA to concentration of 250 ng/μL in sterile PBS buffer. For controls, prepare dsRNA targeting non-endogenous genes (e.g., GFP) at equivalent concentration [1].
- Soaking Treatment: Transfer egg samples to 1.5 mL Eppendorf tubes and immerse in 50 μL dsRNA solution. Incubate at 25°C for 120 minutes to maximize dsRNA uptake [1].
- Post-treatment Handling: Carefully remove dsRNA solution and transfer eggs to appropriate rearing containers with diet.
- Phenotypic Assessment:
- Monitor egg hatching rates daily until completion of embryonic development.
- Assess larval mortality and morphological abnormalities in hatched individuals.
- For molecular validation, extract total RNA from embryos at desired time points using TRIzol reagent and perform qRT-PCR to quantify target gene expression reduction [1].
Chitosan-Nanoparticle Mediated RNAi Delivery
Principle: Chitosan forms stable complexes with dsRNA through electrostatic interactions, protecting it from enzymatic degradation and enhancing cellular uptake, thereby significantly improving RNAi efficiency in recalcitrant insect species [8].
Materials:
- Chitosan (medium molecular weight)
- dsRNA targeting gene of interest (e.g., LmGFAT)
- Sodium tripolyphosphate (TPP) cross-linker
- Magnetic stirrer and heating plate
- Sterile injection equipment or feeding apparatus
Procedure:
- Chitosan-dsRNA Complex Preparation:
- Dissolve chitosan in 1% acetic acid solution to obtain 0.02% (w/v) concentration.
- Prepare dsRNA solution in nuclease-free water at concentration of 200 ng/μL.
- Mix chitosan and dsRNA solutions at optimal N/P ratio (typically 5:1 to 10:1) under gentle stirring at room temperature for 30 minutes to allow complex formation through electrostatic interactions [8].
- Add TPP cross-linker (0.1% w/v) to stabilize nanoparticles.
Delivery Methods:
- Microinjection: Load chitosan-dsRNA complexes into microcapillary needles and inject into insect hemocoel or specific tissues using precision microinjection system [8].
- Oral Feeding: Incorporate chitosan-dsRNA complexes into artificial diet at final dsRNA concentration of 50-100 ng/μL for continuous exposure [8].
Efficacy Assessment:
- Monitor mortality rates daily and record developmental abnormalities.
- Quantify target gene expression at regular intervals post-treatment using RT-qPCR.
- For chitin synthesis targets like GFAT, analyze cuticular chitin content reduction using calorimetric assays or imaging techniques [8].
Signaling Pathways and Molecular Mechanisms
Hormonal Regulation of Vitellogenesis and Embryonic Development
Hormonal Regulation of Insect Reproduction and Embryogenesis: This pathway illustrates the complex interplay between hormone signaling, vitellogenesis, and embryonic development. 20-hydroxyecdysone (20E) and Juvenile Hormone (JH) regulate the nuclear receptor HR3, which directly controls vitellogenin (Vg) and its receptor (VgR) – both critical for yolk deposition and oocyte maturation [12]. Dopamine synthesis enzymes TH and DDC modulate both JH and 20E signaling [10], while 20E additionally regulates chitin biosynthesis [8] and egg hatchability genes [9], creating multiple nodal points for RNAi intervention to disrupt embryonic development.
Experimental Workflow for RNAi-Based Egg Suppression
RNAi Workflow for Embryonic Development Suppression: This comprehensive workflow outlines the systematic approach for developing RNAi-based strategies targeting egg development. Beginning with target gene selection, the process progresses through dsRNA synthesis and delivery optimization using three primary methods: egg soaking, chitosan nanoparticle formulation, and microinjection [1] [8] [11]. The subsequent assessment phase includes phenotypic screening for reduced hatchability and developmental abnormalities, molecular validation of gene silencing efficiency, and final efficacy quantification to determine potential for pest control applications.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for RNAi-Based Egg Development Studies
| Reagent/Resource | Supplier Examples | Application | Technical Considerations |
|---|---|---|---|
| dsRNA Synthesis Kits | Thermo Fisher Scientific, New England Biolabs, Takara Bio | In vitro transcription of high-purity dsRNA | Ensure nuclease-free production; optimize for long fragments (300-500bp) for improved persistence [1] |
| Chitosan Nanoparticles | Sigma-Aldrich, BioSyntan, custom synthesis | dsRNA delivery vector for enhanced cellular uptake and nuclease protection | Optimize N/P ratio for complex formation; particle size <200nm improves tissue penetration [8] |
| Microinjection Systems | Narishige, Drummond, World Precision Instruments | Precise dsRNA delivery into insects or embryos | Calibrate injection volumes (50-100nL) to avoid tissue damage; use fine-tip capillary needles [11] |
| qRT-PCR Reagents | Bio-Rad, Takara, Thermo Fisher | Quantification of gene silencing efficiency | Design primers spanning exon-exon junctions; include reference genes (e.g., RPL32, actin) for normalization [9] |
| Insect Rearing Components | Ward's Science, custom formulations | Maintaining healthy insect colonies for egg production | Standardize artificial diets; control environmental parameters (temperature, humidity, photoperiod) [1] [9] |
RNAi-mediated targeting of vital metabolic and structural proteins represents a promising frontier in pest management by directly undermining egg development and integrity. The protocols and targets outlined herein provide researchers with actionable strategies for disrupting key physiological processes during embryogenesis, from chitin biosynthesis to hormonal regulation. The integration of enhanced delivery platforms, particularly chitosan-based nanoparticles, addresses previous limitations in RNAi efficiency while maintaining environmental compatibility. As the field advances, combination approaches targeting multiple nodes in reproductive pathways may offer synergistic effects for sustainable pest suppression, ultimately reducing reliance on conventional insecticides and their associated ecological impacts.
RNA interference (RNAi) presents a promising biopesticide strategy for agricultural pest control. A primary research objective is the application of RNAi to reduce pest populations by compromising female fecundity and egg hatchability. The success of this approach hinges on the precise identification of essential genes involved in reproduction and embryonic development. This application note details how expression profiling and bioinformatics resources are used to pinpoint such optimal RNAi targets, and provides validated experimental protocols for evaluating their efficacy.
Target Identification: Criteria and Workflow
Effective RNAi targets for reducing fecundity and egg hatchability are typically genes that are highly and specifically expressed in reproductive tissues or during early embryogenesis, and whose silencing leads to significant fitness costs. The following workflow outlines the key steps from gene discovery to functional validation.
Key Identification Criteria:
- High/Focused Expression: Genes with elevated expression in ovaries, fat bodies, or during specific embryonic stages are prioritized [9].
- Essential Function: Genes critical for hormonal signaling, vitellogenesis, eggshell formation, or early embryonic development [1] [9] [10].
- Low Redundancy: Genes with few or non-functional paralogs to prevent functional compensation [13].
- Conservation: Target sequences should be conserved across key pest species but absent in non-target organisms.
Promising Target Genes and Quantitative Outcomes
Recent functional genomics studies have identified several high-value target genes whose silencing drastically reduces egg viability and female fecundity. The quantitative data from these studies are summarized in the table below.
Table 1: Efficacy of Selected RNAi Targets on Pest Reproduction and Survival
| Target Gene | Pest Species | Key Phenotype After RNAi (Egg Hatchability) | Key Phenotype After RNAi (Fecundity) | Key Phenotype After RNAi (Survival/Mortality) | Reference |
|---|---|---|---|---|---|
| Sl102 | Spodoptera littoralis | Drastic reduction in hatching rate; high mortality in hatched larvae [1] | Not specified | Not specified | [1] |
| CYP303A1 | Nilaparvata lugens | Significant reduction in egg hatchability; prolonged embryonic period [9] | No significant effect on ovarian development or oviposition [9] | Not specified | [9] |
| LsTH | Laodelphax striatellus | Inhibited egg hatchability and development [10] | Shortened oviposition period; reduced fecundity [10] | Markedly reduced survival rate [10] | |
| LsDDC | Laodelphax striatellus | Inhibited egg hatchability and development [10] | Shortened oviposition period; reduced fecundity [10] | Markedly reduced survival rate [10] |
The genes listed in Table 1 operate within critical physiological pathways. Silencing them disrupts core processes like the formation of embryonic immune scaffolds and amyloids (Sl102), ecdysteroid biosynthesis and embryonic development (CYP303A1), and dopamine-mediated regulation of juvenile hormone and vitellogenesis (LsTH, LsDDC) [1] [9] [10]. The following diagram illustrates the interconnected signaling pathways affected by these targets.
Experimental Protocol: From dsRNA Design to Egg Hatchability Assay
This section provides a detailed protocol for evaluating the efficacy of a candidate RNAi target, from initial reagent design to a definitive egg hatchability bioassay.
dsRNA Design and In Vitro Synthesis
- Target Sequence Selection: Using a tool like SnapDragon, design a dsRNA sequence (typically 200-500 bp) targeting a specific exon of the candidate gene [13]. Perform specificity checks via BLAST against the pest's transcriptome to avoid off-target effects.
- dsRNA Synthesis: Prepare the template via PCR using gene-specific primers with appended T7 promoter sequences. Synthesize and purify dsRNA using a commercial in vitro transcription kit (e.g., Ambion MEGAscript T7 Kit). Resolve the dsRNA product on an agarose gel to confirm integrity and single-band appearance. Quantify concentration using a spectrophotometer and aliquot for storage at -80°C.
dsRNA Delivery via Egg Soaking
The egg soaking protocol is an effective method for targeting embryonic genes [1].
- Egg Collection: Collect freshly laid egg masses (within 30 minutes of oviposition) from a synchronized pest population. Gently separate individual eggs using a fine brush.
- Soaking Solution Preparation: Prepare a soaking solution containing 250 ng/µL of target-specific dsRNA (e.g., dsSl102) in 1X phosphate-buffered saline (PBS). A dsRNA against an unrelated gene (e.g., GFP) serves as the negative control.
- Soaking Procedure: Place approximately 120 eggs into a 1.5 mL microcentrifuge tube. Submerge them in 50 µL of the dsRNA solution. Incubate for 120 minutes at 25°C ± 1°C [1].
- Post-Treatment Incubation: After soaking, carefully transfer the eggs to a fresh petri dish with a suitable moist substrate. Maintain them under standard insect-rearing conditions until the expected hatching date.
Phenotypic Assessment of Fecundity and Hatchability
- Quantification of Egg Hatching: Daily, record the number of hatched larvae until no further hatching is observed for 48 consecutive hours. Calculate the egg hatching rate as: (Number of Hatched Eggs / Total Number of Treated Eggs) × 100%.
- Larval Mortality Observation: Monitor and record the mortality of the few larvae that do hatch from the dsRNA-treated group [1].
- Fecundity Assay (For Adult-Targeted RNAi): For genes where dsRNA is delivered to adult females (e.g., via injection or feeding), the following should be tracked:
- Pre-oviposition Period: Time from adult emergence to first oviposition.
- Oviposition Period: Total duration of egg-laying.
- Fecundity: Total number of eggs laid per female [10].
Molecular Validation of Silencing
- Sample Collection: Collect a subset of treated eggs at a defined developmental stage (e.g., 32 hours post-oviposition for Sl102) [1].
- RNA Extraction and qRT-PCR: Extract total RNA using TRIzol Reagent. Perform quantitative real-time PCR (qRT-PCR) to measure the relative transcript level of the target gene in dsRNA-treated eggs compared to the control group, confirming successful gene silencing.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Research Tools for RNAi Target Identification and Validation
| Tool / Resource Name | Function / Application | Relevance to RNAi Fecundity Research |
|---|---|---|
| DRscDB [13] | A repository for mining single-cell RNA-seq (scRNA-seq) datasets. | Identifies genes with highly specific expression in ovarian tissues or specific embryonic cell types. |
| DIOPT [13] | Integrative tool for finding orthologs and paralogs across species. | Identifies conserved target genes and assesses potential functional redundancy from paralogs. |
| SnapDragon [13] | Web tool for designing long dsRNA reagents. | Designs effective and specific dsRNA triggers for RNAi experiments in non-model pests. |
| FlyPrimerBank [13] | Database for qPCR primer pairs. | Provides ready-made primers for quantifying gene expression and knockdown efficiency. |
| UP-TORR [13] | Resource for finding Drosophila RNAi transgenic stocks. | Allows rapid functional screening of candidate gene orthologs in the Drosophila model system. |
| TRIzol Reagent [1] [10] | A ready-to-use reagent for total RNA isolation. | Standard method for high-quality RNA extraction from eggs and tissues for downstream transcript analysis. |
| Ambion RETROscript Kit [1] | A complete kit for first-strand cDNA synthesis. | Reverse transcribes RNA into stable cDNA for subsequent qRT-PCR analysis. |
From Lab to Field: Delivery Methods and Application Strategies for Maximum Efficacy
Quantifying Efficacy: Key Performance Metrics from Recent Studies
The tables below summarize quantitative data on the efficacy of soaking and oral dsRNA delivery methods for suppressing embryonic development and reducing fecundity, as reported in recent literature.
Table 1: Efficacy of dsRNA Soaking for Embryonic Silencing
| Insect Species | Target Gene | dsRNA Concentration & Soaking Duration | Key Efficacy Outcomes | Primary Citation |
|---|---|---|---|---|
| Spodoptera littoralis | Sl102 |
250 ng/µL for 120 minutes | Drastic reduction in egg hatching rate; high mortality of hatched larvae; significant developmental delays. | [1] |
| Ostrinia furnacalis | Not Specified | Solution concentration not specified | Effective knockdown leading to developmental retardation and/or death. | [14] |
| Aedes aegypti | Not Specified | Soaking in water containing dsRNA | Effective gene knockdown that persisted into adulthood. | [14] |
| Planarian (S. polychroa) | Various | Soaking in dsRNA solution | Successful gene perturbation in embryos. | [15] |
Table 2: Efficacy of Oral dsRNA Delivery for Reproductive Disruption
| Insect Species | Delivery Method | Target Gene | Key Efficacy Outcomes | Primary Citation |
|---|---|---|---|---|
| Laodelphax striatellus | Oral dsRNA feeding | LsTH (Tyrosine Hydroxylase) |
Shortened oviposition period; reduced fecundity; inhibited egg hatchability and development; reduced survival. | [10] |
| Laodelphax striatellus | Oral dsRNA feeding | LsDDC (Dopa Decarboxylase) |
Shortened oviposition period; reduced fecundity; inhibited egg hatchability and development; reduced survival. | [10] |
| Aethina tumida | Oral feeding (dsRNA-SPc mix) | AtJHAMT (Juvenile Hormone Acid Methyltransferase) |
Impaired ovarian development; reduced JH titers, fecundity, fertility, and egg hatchability. | [16] |
| Bombyx mori (Silkworm) | Oral feeding (Chitosan/dsRNA nanoparticles) | BmToll9-2 (Immune gene) |
Significant gene transcript knockdown; resulted in smaller larvae and cocoons. | [17] |
Detailed Experimental Protocols
Protocol 1: Embryonic Gene Silencing via dsRNA Soaking in Spodoptera littoralis
This protocol, adapted from , details the procedure for suppressing embryonic development in lepidoptera through the soaking of eggs in a dsRNA solution [1].
- Insect Rearing and Egg Collection: Rear adult insects (e.g., Spodoptera littoralis) and allow them to mate. Collect newly laid egg masses within a 30-minute window to ensure a highly synchronized cohort for experimentation. Gently separate individual eggs using a fine brush [1].
- dsRNA Preparation: Synthesize and purify dsRNA targeting your gene of interest (e.g.,
Sl102). As a negative control, prepare dsRNA targeting a non-functional gene such as Green Fluorescent Protein (GFP). Resuspend the dsRNA in 1X Phosphate Buffered Saline (PBS) at the desired working concentration (e.g., 50, 100, or 250 ng/µL) [1]. - Soaking Procedure: Place approximately 120 synchronized eggs into a 1.5 mL microcentrifuge tube. Add 50 µL of the dsRNA solution to completely submerge the eggs. Incubate the tube at 25 ± 1 °C for the desired duration (e.g., 30, 60, or 120 minutes) to allow for dsRNA uptake [1].
- Post-Treatment Incubation and Hatching Assessment: After soaking, carefully transfer the eggs to a standard diet or appropriate growth medium. Maintain them under controlled environmental conditions (e.g., 25 °C, 70% RH, 16:8 light/dark photoperiod). Record the number of hatched eggs to calculate the hatching rate. Monitor the few hatched larvae for mortality and morphological alterations [1].
- Efficacy Validation (qRT-PCR): To confirm gene silencing, extract total RNA from a subset of treated eggs at a specific developmental time point using TRIzol Reagent. Perform absolute quantitative Real-Time PCR (qRT-PCR) to measure the transcript levels of the target gene relative to control eggs [1].
Protocol 2: Oral RNAi in Lepidopteran Larvae Using Chitosan/dsRNA Nanoparticles
This protocol, based on , describes the formulation of chitosan/dsRNA nanoparticles for effective oral RNAi in insects like the silkworm, Bombyx mori, where naked dsRNA is ineffective due to gut nucleases [17].
- Nanoparticle Formulation: Prepare a 0.02% (w/v) chitosan solution in sodium acetate buffer (pH 5.5). Mix the chitosan solution with an equal volume of dsRNA solution (targeting a gene of interest, e.g.,
BmToll9-2) under constant vortexing for 30 seconds. Allow the mixture to self-assemble for at least 2 hours at room temperature to form stable chitosan/dsRNA nanoparticles. Characterize the resulting nanoparticles using Transmission Electron Microscopy (TEM) and dynamic light scattering to confirm their spherical morphology and size (~80 nm) [17]. - Oral Delivery via Feeding: For silkworms, administer the nanoparticles by coating them onto fresh mulberry leaves. For other insects, the nanoparticle solution can be incorporated into an artificial diet. Allow the insects to feed ad libitum on the treated food source. To maintain a consistent RNAi effect, re-administer the nanoparticles every 3-5 days, as the effect is transient [17].
- Phenotypic and Molecular Analysis: Monitor and record phenotypic changes such as larval growth reduction, developmental defects, or mortality. For molecular confirmation, dissect midgut or other relevant tissues from treated larvae. Extract total RNA and synthesize cDNA. Use quantitative RT-PCR to quantify the knockdown efficiency of the target gene transcript compared to control groups fed with naked dsRNA or dsRNA targeting a neutral gene [17].
Visualizing Experimental Workflows
dsRNA Soaking and Oral Delivery Pathways
The diagram below illustrates the core workflows for inducing RNAi via soaking and oral delivery, highlighting the critical role of nanoparticle protection.
Dopamine Synthesis as a Target for Reproductive Disruption
Targeting the dopamine synthesis pathway through oral RNAi is an effective strategy to impair insect reproduction, as shown in Laodelphax striatellus [10].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for dsRNA-Mediated Embryonic and Reproductive Silencing
| Reagent / Tool | Function / Application | Key Characteristics & Examples |
|---|---|---|
| Chitosan Nanoparticles | Protects dsRNA from degradation in the insect gut; facilitates cellular uptake. | Biodegradable, cationic polymer. Used for oral RNAi in lepidoptera (e.g., Bombyx mori) [17]. |
| Cell-Penetrating Peptides (CPPs) | Enhances cellular internalization of dsRNA. | e.g., PTD-DRBD (Peptide Transduction Domain - dsRNA Binding Domain). Forms ribonucleoprotein particles (RNPs) that shield dsRNA and improve uptake [18]. |
| Gold Nanoparticles (AuNPs) | Versatile platform for oligonucleotide delivery; can be functionalized with targeting ligands. | High functionalization capacity, low toxicity. Can be conjugated with aptamers (e.g., against α7/β1 integrin) for targeted delivery [19]. |
| Target Genes for Reproductive Disruption | Genes whose silencing leads to reduced fecundity, egg hatchability, or embryonic development. | - TH & DDC: Key enzymes in dopamine synthesis, essential for reproduction in Laodelphax striatellus [10].- JHAMT: Critical for juvenile hormone synthesis; silencing impairs ovarian development in Aethina tumida [16].- Sl102: Involved in immune response and basal lamina formation; silencing disrupts embryonic development in Spodoptera littoralis [1]. |
| Chemical Modifications | Increases dsRNA stability against nuclease degradation. | Phosphorothioate (PS) backbone modifications; 2'-O-Me, 2'-O-Et, or 2'-F ribose substitutions; Locked Nucleic Acid (LNA) [20]. |
RNA interference (RNAi) has emerged as a promising, eco-friendly alternative to chemical pesticides for pest management in agriculture [21] [22]. This gene silencing technique functions by introducing double-stranded RNA (dsRNA) into pest organisms, which triggers a sequence-specific degradation of complementary messenger RNA (mRNA), disrupting the expression of essential genes [22]. Transgenic plant systems that express pest-targeted dsRNA represent a sustainable and self-delivering platform for dsRNA production [23]. When framed within research aimed at reducing pest fecundity and egg hatchability, this technology offers a powerful strategy for suppressing pest populations at their earliest developmental stages [1]. These Application Notes and Protocols detail the design, production, and efficacy testing of dsRNA for targeting pest reproductive and embryonic genes.
Key dsRNA Design Parameters for Targeting Fecundity and Embryonic Development
The efficacy of RNAi hinges on the rational design of the dsRNA molecule. Beyond selecting an essential target gene, parameters such as length, sequence features, and secondary structure must be optimized to maximize gene silencing and the resulting phenotypic effects, such as reduced egg hatchability.
Table 1: Key Parameters for Optimizing dsRNA Design for Insecticidal Activity
| Parameter | Optimal Characteristic | Biological Rationale | Empirical Support |
|---|---|---|---|
| dsRNA Length | >60 bp; typically 200-500 bp | Longer dsRNAs enable more efficient cellular uptake and are processed into multiple siRNAs, increasing the likelihood of effective silencing [22]. | In Tribolium castaneum, longer dsRNAs were more effective in silencing specific genes [21] [22]. |
| Thermodynamic Asymmetry | Weak binding at the 5' end of the antisense (guide) strand | Promotes preferential loading of the antisense strand into the RISC complex, guiding it to the target mRNA [21]. | A key predictor of high efficacy in T. castaneum; associated with higher ratio of antisense siRNA in RISC [21]. |
| Nucleotide Preference | Adenine at the 10th position of the antisense siRNA | Correlates with high insecticidal efficacy, though the precise mechanistic role is under investigation [21]. | Identified as a predictive feature for efficacy in systematic screens in T. castaneum [21]. |
| GC Content (nucleotides 9-14) | High GC content | In contrast to human data, high GC in this region of the antisense strand was associated with high efficacy in insects [21]. | Empirical finding in T. castaneum, differing from canonical design rules based on human cells [21]. |
| Secondary Structures | Absence of stable secondary structures in target mRNA | Accessible mRNA regions without complex folding are more susceptible to RISC binding and cleavage [21]. | A predictive feature for high RNAi efficacy [21]. |
The gene Sl102 in Spodoptera littoralis serves as a prime example of a viable target for reducing egg hatchability. This gene encodes a protein involved in forming functional amyloid fibrils crucial for immune response and embryonic development, including the formation of the basal lamina in epithelial tissues [1]. Silencing Sl102 during embryogenesis causes significant developmental delays, morphological alterations, and drastically reduces the egg hatching rate, complemented by high mortality of the few larvae that do hatch [1].
Experimental Protocols
Protocol 1: In Vitro Screening of dsRNA Efficacy via Egg Soaking
This protocol is adapted from successful RNAi induction in embryos of Spodoptera littoralis and other insects [1]. It allows for rapid, high-throughput screening of candidate dsRNAs targeting genes involved in fecundity and embryonic development before moving to plant transformation.
Workflow Overview:
Materials & Reagents:
- dsRNA: Targeting the gene of interest (e.g.,
Sl102) and a non-target control (e.g.,GFP). Produce via bacterial expression systems (see Protocol 2) or commercial in vitro transcription kits. - Insect Strain: Synchronized egg masses from the target pest species.
- Soaking Buffer: 1X Phosphate Buffered Saline (PBS).
- Equipment: Sterile microcentrifuge tubes, precision pipettes, environmental chamber for insect rearing.
Procedure:
- dsRNA Production: Synthesize and purify dsRNA. Quantify concentration using a spectrophotometer and verify integrity via agarose gel electrophoresis.
- Egg Collection: Collect newly laid egg masses (within a 30-minute window) from an adult colony. Gently separate individual eggs with a soft brush to ensure uniform treatment [1].
- Soaking Treatment:
- Prepare a 50 µL soaking solution of dsRNA in PBS. A concentration of 250 ng/µL is effective for many systems, but a dose-response curve (e.g., 50, 100, 250 ng/µL) is recommended for optimization [1].
- Transfer approximately 120 synchronized eggs to a 1.5 mL microcentrifuge tube containing the dsRNA solution.
- Soak the eggs for 120 minutes at 25 ± 1 °C, gently agitating periodically to ensure full immersion [1].
- For the control group, treat an equivalent number of eggs with dsRNA targeting a non-functional gene (e.g.,
GFP).
- Post-Treatment Incubation: After soaking, carefully remove the solution. Transfer the eggs to a fresh Petri dish with a suitable substrate and incubate them under standard conditions (e.g., 25 °C, 70% RH) until the control group hatches.
- Phenotypic Assessment:
- Record the number of hatched eggs daily to calculate the hatching rate.
- Observe and document any morphological abnormalities in the embryos or hatched larvae.
- Monitor larval mortality post-hatching.
- Molecular Validation: To confirm gene silencing, extract total RNA from treated and control eggs at a specific developmental stage (e.g., 32 hours after oviposition for
Sl102). Perform absolute quantitative RT-PCR to measure the relative transcript levels of the target gene [1].
Protocol 2: Sustainable dsRNA Production in Transgenic Plants
This protocol outlines the process from target selection to the generation of transgenic plants that continuously produce pest-targeted dsRNA.
Workflow Overview:
Materials & Reagents:
- Target Sequence: A ~200-500 bp fragment from the pest target gene (e.g.,
Sl102), optimized using parameters in Table 1. - Plant Binary Vector: A vector containing an inverted repeat sequence compatible with plant transcription, separated by an intron spacer to facilitate dsRNA formation after splicing.
- Plant Material: Model or crop species (e.g., Nicotiana benthamiana, tomato) amenable to genetic transformation.
- Stable Transformation Reagents: Agrobacterium tumefaciens strain, tissue culture media, selective agents (e.g., antibiotics).
Procedure:
- Target Gene & dsRNA Region Selection:
- Identify essential genes for pest reproduction or embryogenesis (e.g.,
Sl102). - Using a platform like dsRIP, select a 200-500 bp fragment within the target mRNA that is predicted to yield highly effective siRNAs based on insect-specific features (thermodynamic asymmetry, specific nucleotide preferences, etc.) [21].
- Identify essential genes for pest reproduction or embryogenesis (e.g.,
- Plant Transformation Vector Construction:
- Synthesize the selected DNA fragment and clone it as an inverted repeat into a plant binary vector under the control of a constitutive plant promoter (e.g., Cauliflower Mosaic Virus 35S promoter).
- The construct should include a plant-selectable marker gene (e.g., for kanamycin resistance).
- Plant Transformation:
- Introduce the constructed vector into Agrobacterium tumefaciens.
- Transform the plant of choice using standard methods for that species, such as the floral dip method for Arabidopsis or Agrobacterium-mediated transformation of leaf discs for solanaceous crops.
- Regenerate transformed plants on selective media.
- Molecular Characterization of Transgenics:
- Confirm the integration of the transgene in primary transformants (T0 plants) by PCR.
- Analyze dsRNA expression in T1 or subsequent generations using techniques like RT-PCR or northern blotting.
- Bioefficacy Testing:
- Use leaf material from transgenic plants to feed adults or larvae of the target pest and assess fecundity (number of eggs laid) and fertility (egg hatchability) [1].
- Compare results to pests fed on non-transformed control plants or plants expressing a non-target dsRNA.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Materials for dsRNA-based Pest Control Research
| Item | Function/Application | Examples & Notes |
|---|---|---|
| dsRIP Web Platform | A specialized tool for designing optimized dsRNA sequences, identifying effective target genes in pests, and assessing risks to non-target species [21]. | Publicly available platform incorporating insect-specific siRNA features (e.g., high GC from 9-14th nt) for rational design [21]. |
| Bacterial dsRNA Production System | Cost-effective, scalable production of dsRNA for high-throughput screening and topical applications [24]. | E. coli HT115 (DE3) with RNase III deficiency, often using the L4440 vector [24]. |
| RNA Isolation Kits | High-quality dsRNA purification from bacterial or plant tissue with high yield and purity. | TRIzol-absolute ethanol method yields high total RNA; ethanol isolation offers superior dsRNA recovery efficiency (~84%) [24]. |
| Virus-Induced Gene Silencing (VIGS) Vectors | A transient, non-transgenic method for rapid functional validation of target genes directly in plants or for pest control [25]. | vsRNAi technique uses viral vectors with ultra-short RNA inserts (24 nt) for highly specific gene silencing [25]. |
| In Vitro Transcription Kits | Rapid synthesis of small quantities of dsRNA for initial, small-scale bioassays. | Useful for generating dsRNA for egg soaking assays without needing a bacterial system. |
| Lipid Nanoparticles (LNPs) | A delivery system to protect dsRNA from environmental degradation and enhance cellular uptake in sprayable formulations [26]. | Leading delivery system in RNAi therapeutics; shows promise for enhancing foliar applications in agriculture [26]. |
Transgenic plant systems for the sustainable production of dsRNA represent a cutting-edge strategy within integrated pest management. By focusing on molecular design parameters that enhance RNAi efficacy—such as thermodynamic asymmetry and insect-specific nucleotide preferences—researchers can develop highly effective plant-based solutions. Targeting genes critical for fecundity and embryonic development, like Sl102, offers a pathway to suppress pest populations at the egg stage, preventing crop damage before it begins. The protocols and tools provided here offer a roadmap for developing and testing these innovative pest control solutions.
The application of RNA interference (RNAi) to suppress insect populations by reducing fecundity and egg hatchability represents a promising frontier in pest management and vector control. The core challenge in realizing this strategy lies in the efficient delivery of RNAi triggers, such as double-stranded RNA (dsRNA), to the target insect tissues. This document details application notes and protocols for two primary delivery strategies—viral vectors and nanoparticle formulations—framed within the context of this research goal. The success of these approaches hinges on their ability to overcome significant biological barriers, including dsRNA degradation by nucleases, inefficient cellular uptake, and endosomal entrapment, to achieve effective gene silencing [27] [1] [28].
Comparative Analysis of Delivery Platforms
The choice of delivery system is critical. The table below summarizes the key characteristics of viral and non-viral platforms for delivering RNAi effectors in insect systems.
Table 1: Comparison of Delivery Platforms for RNAi in Fecundity and Hatchability Research
| Feature | Viral Vectors (e.g., LV, Ad, AAV) | Non-Viral Vectors (e.g., Lipid Nanoparticles) |
|---|---|---|
| Core Mechanism | Use natural viral infection pathways for high-efficiency delivery [27] [29]. | Package and protect dsRNA/mRNA; facilitate cellular uptake through engineered lipids and polymers [27] [30]. |
| Transfection Efficiency | Typically high [27] [29]. | Variable; often lower than viral vectors but continuously improving [27]. |
| Cargo Capacity | Limited (e.g., AAV: ~4.5 kb) [31] [30]. | Higher capacity, suitable for large dsRNA constructs [30]. |
| Immunogenicity | Can be immunogenic, potentially triggering host immune responses [27] [31]. | Generally lower immunogenicity, but can still induce inflammatory responses [27] [30]. |
| Production Complexity | Complex and costly [27]. | Simpler, more scalable, and cost-effective [27] [30]. |
| Safety Profile | Risks associated with pre-existing immunity and insertional mutagenesis (for some classes) [31] [30]. | Safer profile; no risk of genomic integration [30]. |
| Key Application in RNAi | Suitable for long-term or systemic gene silencing studies in model insects [32] [33]. | Ideal for topical applications (e.g., spray-induced gene silencing) and oral delivery via soaked bait [1] [26]. |
Quantitative Efficacy Data
The following table compiles quantitative data from key studies that successfully utilized these delivery platforms to suppress fecundity and egg hatchability in various insect species.
Table 2: Efficacy Metrics of Delivery Platforms in Reducing Fecundity and Hatchability
| Target Insect / Gene | Delivery Platform & Method | Key Efficacy Metrics | Reference |
|---|---|---|---|
| Spodoptera littoralis / Sl102 | Non-viral: Soaking eggs in dsRNA solution (250 ng/µL for 120 min) [1]. | - Drastic reduction in egg hatching rate.- Very high mortality of the few hatched larvae [1]. | [1] |
| Aedes aegypti / LAP1 | Viral (CRISPR/Cas9): CRISPR/Cas9-mediated deletion to create LAP1⁻/⁻ mutant males [32]. | - Reduction in reproduction when wild-type females mated with LAP1⁻/⁻ males [32]. | [32] |
| Aedes aegypti / LAP1, M12 | Non-viral (RNAi): Knockdown of genes via dsRNA injection in females [32]. | - Suppression of both fecundity (egg deposition) and fertility (hatchability) in LAP1 and M12 dsRNA-treated mosquitoes [32]. | [32] |
| General Lepidoptera | Non-viral: Microinjection of dsRNA into embryos [34]. | - Established protocol for effective gene knockdown in embryos, a sensitive life stage [34]. | [34] |
Experimental Protocols
Protocol: Parental RNAi via Egg Soaking inSpodoptera littoralis
This protocol describes a method for suppressing embryonic development by soaking eggs in a dsRNA solution, targeting genes essential for fecundity and hatchability [1].
1. Research Reagent Solutions
Table 3: Essential Reagents for Egg Soaking Protocol
| Item | Function / Description |
|---|---|
| dsRNA (target gene) | The effector molecule for RNAi; designed against a target gene (e.g., Sl102). |
| dsRNA (control) | Control dsRNA targeting a non-functional gene (e.g., GFP). |
| PBS (1X) | Phosphate Buffered Saline; the physiological buffer used to deliver and soak dsRNA. |
| TRIzol Reagent | For subsequent total RNA extraction from eggs to validate gene silencing. |
2. Step-by-Step Workflow
- Step 1: Insect Rearing and Egg Collection. Rear S. littoralis larvae on an artificial diet under controlled conditions (e.g., 25 ± 1 °C, 70 ± 5% RH). Collect newly laid egg masses from adults and separate individual eggs with a fine brush to create a synchronized experimental group [1].
- Step 2: dsRNA Preparation. Synthesize and purify dsRNA targeting your gene of interest (e.g., Sl102) and a control dsRNA (e.g., dsGFP). Resuspend the dsRNA in nuclease-free 1X PBS to the desired working concentration (e.g., 50-250 ng/µL) [1].
- Step 3: Egg Soaking Treatment.
- Collect approximately 120 synchronized eggs in a 1.5 mL microcentrifuge tube.
- Soak the eggs in 50 µL of the dsRNA solution (e.g., 250 ng/µL).
- Incubate the tube at 25 ± 1 °C for the determined duration (e.g., 30, 60, or 120 minutes) [1].
- Step 4: Post-Treatment Incubation and Data Collection.
- After soaking, carefully transfer the eggs to a fresh diet or suitable moist substrate.
- Maintain the eggs under standard rearing conditions.
- Record the number of hatched eggs to calculate the hatching rate.
- For the few hatched larvae, monitor and record larval mortality [1].
- Step 5: Validation of Gene Silencing (qRT-PCR).
- Extract total RNA from a subset of treated eggs at a specific time point post-soaking using TRIzol Reagent.
- Perform reverse transcription and quantitative real-time PCR (qRT-PCR) to measure the transcript level of the target gene relative to control genes. Successful silencing is confirmed by a significant reduction in target gene mRNA [1].
Diagram 1: Egg Soaking RNAi Workflow
Protocol: Formulation of Lipid Nanoparticles (LNPs) for dsRNA Delivery
This protocol outlines the formulation of LNPs, a leading non-viral delivery system, for encapsulating and protecting dsRNA [27] [26] [30].
1. Research Reagent Solutions
Table 4: Essential Reagents for LNP Formulation
| Item | Function / Description |
|---|---|
| Ionizable Cationic Lipid | Critical for endosomal escape; protonated in acidic endosomes, disrupting the membrane [27]. |
| Helper Lipid (e.g., DOPE, DSPC) | Stabilizes the LNP structure and supports membrane fusion. |
| Cholesterol | Enhances the stability and rigidity of the LNP bilayer. |
| PEG-lipid (e.g., DMG-PEG2000) | Shields the LNP surface, reduces aggregation, and modulates pharmacokinetics. |
| dsRNA payload | The therapeutic agent to be encapsulated. |
2. Step-by-Step Workflow
- Step 1: Preparation of Lipid Mixture. Dissolve the ionizable lipid, helper phospholipid, cholesterol, and PEG-lipid in ethanol at a specific molar ratio. The composition can be optimized for insect cell delivery [27].
- Step 2: Preparation of Aqueous Phase. Dilute the dsRNA in an acidic aqueous buffer (e.g., citrate buffer, pH 4.0). This facilitates the electrostatic interaction between the negatively charged dsRNA and the protonatable cationic lipid [27].
- Step 3: Nanoparticle Formation. Rapidly mix the ethanolic lipid solution with the aqueous dsRNA solution using a microfluidic device or rapid pipetting. This process leads to the spontaneous formation of LNPs encapsulating the dsRNA [27] [30].
- Step 4: Buffer Exchange and Purification. Dialyze or use tangential flow filtration to exchange the LNP suspension into a neutral, physiological buffer (e.g., PBS) to remove ethanol and stabilize the particles for storage and application.
- Step 5: Characterization. Determine the particle size and polydispersity (PDI) using dynamic light scattering. Measure the encapsulation efficiency of the dsRNA using a dye exclusion assay [27].
Diagram 2: LNP Formulation Process
The Scientist's Toolkit: Key Research Reagents
Table 5: Essential Materials for RNAi-based Fecundity Research
| Category / Item | Specific Examples | Function in Research |
|---|---|---|
| Delivery Vectors | ||
| ∙ Viral Vectors | Lentivirus (LV), Adenovirus (Ad), Adeno-associated virus (AAV) [29] [33]. | Engineered for high-efficiency gene delivery and long-term silencing in model insects. |
| ∙ Non-Viral Vectors | Lipid Nanoparticles (LNPs), Polymeric Nanoparticles [27] [26]. | Protect dsRNA, enhance cellular uptake, and can be used in topical or oral delivery strategies. |
| RNAi Triggers | ||
| ∙ dsRNA | In vitro transcribed dsRNA [1] [34]. | The direct effector molecule for initiating the RNAi pathway. |
| ∙ siRNA | Chemically synthesized siRNA [26]. | Defined, short RNA duplexes; offer high specificity. |
| Formulation Components | ||
| ∙ Cationic/Ionizable Lipids | DOTAP, DLin-MC3-DMA [27]. | Bind nucleic acids and facilitate endosomal escape. |
| ∙ Polymers | Polyethyleneimine (PEI), Chitosan (CS) [30]. | Condense nucleic acids into polyplexes for delivery. |
| ∙ Targeting Ligands | GalNAc (for hepatocytes), peptide ligands [26]. | Can be conjugated to nanoparticles to enhance target cell specificity. |
| Analytical Tools | ||
| ∙ Gene Expression | qRT-PCR reagents [1]. | Quantify knockdown efficiency of the target gene. |
| ∙ Phenotypic Assays | Hatching rate count, larval mortality tracking, fecundity assessment (eggs/female) [1] [32]. | Measure the functional biological outcome of gene silencing. |
Application Notes
High-Throughput RNAi Screening for Embryonic Development Genes
This application note details a high-throughput screening (HTS) approach to identify genes critical for fecundity and egg hatchability in Lepidoptera, specifically Spodoptera littoralis, using RNAi technology. The primary objective was to discover target genes whose suppression disrupts embryonic development, providing a novel pest control strategy. The gene Sl102, which encodes precursors of functional amyloid fibrils, was screened and identified as a key regulator. The screening protocol involved soaking highly synchronized eggs in dsRNA solutions, enabling high-throughput processing of numerous samples to assess the impact on hatching rates and larval mortality [1].
Key Quantitative Outcomes from RNAi Screening of Sl102: The table below summarizes the core experimental findings, demonstrating that prolonged exposure to higher dsRNA concentrations drastically reduces viable offspring.
| Target Gene | dsRNA Concentration (ng/µL) | Soaking Duration (min) | Reduction in Egg Hatching Rate | Mortality of Hatched Larvae |
|---|---|---|---|---|
| Sl102 | 250 | 120 | Drastic Reduction [1] | Very High [1] |
| RpL11 | Information Not Specified | Information Not Specified | 20.4% [35] | Information Not Specified |
| RpS2 | Information Not Specified | Information Not Specified | 22.4% [35] | Information Not Specified |
| tra-2 | Information Not Specified | Information Not Specified | 30.6% [35] | Information Not Specified |
Impact on Oviposition and Development: Beyond hatching success, RNAi-mediated silencing of ribosomal proteins RpL11 and RpS2 in other species has been shown to cause a significant reduction in fecundity and oviposition duration, indicating their role in egg formation [35]. Ultrastructural and morphological analyses of Sl102-silenced embryos revealed significant developmental delays and alterations, confirming a vital role in embryonic development [1].
Automation and Workflow Integration for Enhanced Screening
The adoption of HTS methodologies is revolutionizing the efficiency of biological research and drug discovery. The global HTS market is projected to grow from USD 26.12 billion in 2025 to USD 53.21 billion by 2032, reflecting its critical role [36]. A demonstrated HTS workflow for nearly 10,000 protein samples achieved an 80% reduction in hands-on time by leveraging a fully automated Reconfigurable Automation Cart (RAC) platform, enabling data generation without in-person monitoring [37]. This level of automation is crucial for the scalable application of RNAi screening protocols.
Key High-Throughput Screening Market Drivers: The following table breaks down the dominant segments and regional markets, highlighting the technologies and areas with the highest growth and adoption.
| Segment | Projected Market Share (2025) | Key Drivers and Technologies |
|---|---|---|
| Overall HTS Market | $26.12 Billion [36] | Faster drug discovery, automation, AI integration [36] |
| Product & Services | Instruments (Liquid Handlers, Readers): 49.3% [36] | Automation, precision, miniaturization (nanoliter scales) [36] |
| Technology | Cell-Based Assays: 33.4% [36] | Demand for physiologically relevant models [36] |
| Application | Drug Discovery: 45.6% [36] | Need for rapid, cost-effective candidate identification [36] |
| Region | North America: 39.3% (Leader) [36] | Advanced infrastructure, major industry players, funding [36] |
| Region | Asia Pacific: 24.5% (Fastest Growing) [36] | Expanding pharma industries, rising R&D investments [36] |
Experimental Protocols
Protocol 1: High-Throughput RNAi Screening via Egg Soaking for Fecundity and Hatchability Studies
This protocol describes a robust method for inducing RNAi in insect eggs through dsRNA soaking, adapted for high-throughput screening of genes affecting embryogenesis, fecundity, and egg hatching [1].
Materials and Reagents
- Insects: Highly synchronized, newly laid eggs (e.g., from Spodoptera littoralis).
- dsRNA: Target-specific dsRNA (e.g., Sl102, RpL11, RpS2) and control dsRNA (e.g., dsGFP).
- Buffers: 1X Phosphate Buffered Saline (PBS): 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4; pH 7.4.
- Equipment: Microcentrifuge tubes, precision pipettes, controlled temperature chamber (25 ± 1 °C).
Procedure
Egg Collection and Preparation:
- Collect newly laid eggs (e.g., 120 eggs) within a narrow time window (e.g., 30 minutes) to ensure developmental synchronization [1].
- Gently place the eggs in a 1.5 mL microcentrifuge tube.
dsRNA Soaking Treatment:
- Prepare the dsRNA soaking solution by diluting the target or control dsRNA in 1X PBS to the desired concentration (e.g., 50, 100, or 250 ng/µL). A volume of 50 µL is sufficient for 120 eggs [1].
- Add the dsRNA solution to the tube containing the eggs, ensuring they are fully immersed.
- Incubate the tube at 25 ± 1 °C for the desired duration (e.g., 30, 60, or 120 minutes) [1].
Post-Treatment Incubation and Data Collection:
- Following the soaking period, carefully remove the solution.
- Transfer the eggs to a suitable container for incubation under standard conditions (e.g., 25 °C, 70% RH) until the expected hatching period.
- Phenotypic Assessment:
- Hatching Rate: Daily record the number of hatched eggs.
- Larval Mortality: Monitor and record the mortality of hatched larvae.
- Fecundity Assessment (For RNAi on adults): For genes tested in adult females (e.g., RpL11), track the number of eggs laid (fecundity) and the subsequent hatching rate [35].
Molecular Validation (qRT-PCR):
Data Analysis
- Calculate the percentage reduction in hatching rate and fecundity in dsRNA-treated groups compared to the control group.
- Determine the mortality rate of hatched larvae.
- Analyze qRT-PCR data to confirm the correlation between phenotypic effects and the level of target gene suppression.
Protocol 2: High-Throughput Fluorescent-Based Screening for Enzyme Inhibitors
This protocol provides a framework for high-throughput screening of enzymatic activity and inhibitors, adaptable for targeting enzymes involved in reproductive biology. The example given is for screening SIRT7 inhibitors but illustrates a universally applicable HTS methodology [38].
Materials and Reagents
- Recombinant Protein: Purified target enzyme (e.g., His-tagged SIRT7).
- Substrate: Fluorescently-labeled peptide substrate specific to the target enzyme.
- Compound Library: A library of chemical compounds for inhibitor screening.
- Assay Plates: 384-well microplates suitable for fluorescence measurements.
- Detector: A fluorescent plate reader.
Procedure
- Protein Purification: Purify a recombinant, active form of the target protein (e.g., His-SIRT7 from E. coli) for use in the assay [38].
- Enzymatic Reaction Setup:
- In each well of the microplate, combine the purified enzyme, the fluorescent peptide substrate, and a single compound from the library.
- Include positive (enzyme + substrate without inhibitor) and negative controls (substrate only).
- Reaction Incubation and Signal Detection:
- Allow the enzymatic reaction to proceed under optimized conditions (e.g., specific temperature, time).
- Measure the change in fluorescent signal using a microplate reader configured for the appropriate excitation/emission spectrum [38].
- Hit Identification and Validation:
- Identify "hits" as compounds that significantly reduce the fluorescent signal compared to the positive control.
- For confirmed hits, perform dose-response curves to determine the half-maximal inhibitory concentration (IC50) [38].
Signaling Pathways and Workflows
RNAi Mechanism for Gene Suppression
This diagram illustrates the core mechanism of RNA interference (RNAi) leading to the suppression of a target gene, a process central to the protocols described.
High-Throughput RNAi Screening Workflow
This diagram outlines the complete end-to-end workflow for a high-throughput RNAi screen targeting egg hatchability and fecundity.
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Reagents for RNAi-based High-Throughput Screening
| Research Reagent | Function in the Protocol |
|---|---|
| Target-specific dsRNA | The core reagent; a double-stranded RNA molecule designed to be complementary to the target messenger RNA (mRNA), triggering its degradation and silencing the gene of interest [1]. |
| Control dsRNA (e.g., dsGFP) | A critical negative control; a dsRNA molecule with no target in the experimental organism, used to account for non-specific effects of the dsRNA delivery process [1]. |
| Synchronized Insect Eggs | Biologically relevant assay subjects; eggs laid within a very short time window ensure uniform developmental stages, which is essential for reproducible and interpretable high-throughput screening results [1]. |
| Fluorescent Peptide Substrate | Enables activity measurement; a peptide linked to a fluorescent group used in enzymatic assays to track enzyme activity via fluorescence change, facilitating high-throughput inhibitor screening [38]. |
| Liquid Handling Systems | Enables automation; automated instruments that precisely dispense nanoliter to microliter volumes of reagents (dsRNA, buffers) into multi-well plates, crucial for speed, accuracy, and scalability in HTS [36]. |
| Microplate Readers (Detectors) | Detects assay outputs; instruments that measure signals (e.g., fluorescence, luminescence) from multi-well plates, allowing for the high-speed quantitative data capture required in HTS [36] [38]. |
Overcoming Hurdles: Strategies for Enhancing RNAi Efficiency and Specificity
A major barrier to achieving consistent RNA interference (RNAi), particularly in research aimed at reducing insect fecundity and egg hatchability, is the rapid degradation of double-stranded RNA (dsRNA) before it can reach its target cells. Environmental factors like ultraviolet light and ubiquitous nucleases in soil, insect hemolymph, gut, and saliva efficiently cleave and inactivate dsRNA molecules [39] [40]. This degradation severely compromises the efficacy of RNAi-based strategies, leading to variable experimental results and insufficient gene silencing. This Application Note details proven methodologies combining chemical modifications and nuclease inhibition strategies to protect dsRNA, thereby enhancing the reliability and potency of RNAi applications in pest control and functional genomics.
Strategic Approaches to dsRNA Protection
Two primary, complementary strategies exist to safeguard dsRNA integrity: modifying the dsRNA molecule itself to increase its inherent stability, and using formulations that inhibit or evade degradative nucleases. Figure 1 illustrates the core challenges and the strategic solutions detailed in this note.
Figure 1. Strategic overview of combating dsRNA degradation. The primary challenges (left) lead to degraded dsRNA and poor RNAi outcomes. The two core solution strategies (right)—chemical modification and nuclease inhibition—work synergistically to protect dsRNA and ensure high RNAi efficiency.
Chemical Modifications of dsRNA
Chemical modification of the dsRNA backbone and sugar moieties is a fundamental approach to confer resistance against nuclease attack. These modifications are designed to impair nuclease binding and cleavage without disrupting the dsRNA's ability to engage the RNAi machinery [20].
Table 1: Common Chemical Modifications for Enhancing dsRNA Stability
| Modification Type | Chemical Structure | Key Function | Effect on Stability & Activity |
|---|---|---|---|
| Phosphorothioate (PS) Backbone | Replaces non-bridging oxygen with sulfur in phosphate backbone [20]. | Reduces hydrolysis by nucleases; improves binding to plasma proteins [20]. | Increased nuclease resistance; may slightly reduce binding affinity. |
| 2'-Sugar Modifications (2'-O-Me, 2'-O-Et, 2'-F) | Replaces the 2'-hydroxyl group (2'-OH) of ribose with -O-methyl, -O-ethyl, or fluorine [20]. | Sterically hinders RNase binding; critical for reducing immunogenicity [20]. | Dramatically increased stability in serum/hemolymph; maintained activity within RISC. |
| Locked Nucleic Acid (LNA) | Additional methylene bridge between 2'-oxygen and 4'-carbon, "locking" the ribose [20]. | Greatly improved base-pairing affinity (hybridization) and specificity [20]. | Enhanced thermal stability and nuclease resistance; requires careful design to avoid hepatotoxicity in therapeutics [20]. |
Nuclease Inhibition & Advanced Formulations
Beyond altering the dsRNA itself, formulating it with protective carriers or competitive inhibitors provides a powerful physical and biochemical shield against degradation.
Nanoparticle-Based Delivery Systems
Nanocarriers protect dsRNA via encapsulation and enhance cellular uptake. Figure 2 outlines a general workflow for preparing and testing nanoparticle-dsRNA complexes.
Figure 2. Experimental workflow for nano-enabled dsRNA formulation. The process involves creating nanoparticles, complexing them with dsRNA, characterizing the complexes, and rigorously testing their stability and efficacy.
Table 2: Nanoparticle Systems for dsRNA Delivery and Nuclease Protection
| Nanocarrier System | Composition & Formation | Key Protective Mechanism | Documented Efficacy |
|---|---|---|---|
| ZIF-8@PDA | Zeolitic Imidazolate Framework-8 core with a Polydopamine shell; self-assembled with dsRNA [41]. | Protects from enzymatic hydrolysis in gut fluid (GF) and hemolymph (HL); enhances cellular uptake via endocytosis [41]. | 357.9-fold higher fluorescence intensity in Sf9 cells vs. naked dsRNA; significant increase in insect mortality [41]. |
| Chitosan-based | Natural polysaccharide; forms polyplexes with dsRNA via electrostatic interaction [8] [39]. | Protects dsRNA from nucleases in alkaline gut environments; improves penetration of the peritrophic matrix [8] [39]. | dsLmGFAT complexed with chitosan led to ~90% mortality in Locusta migratoria, vs. 70% with naked dsRNA [8]. |
| ε-PL@CMCS | ε-poly-L-lysine and carboxymethyl chitosan self-assembled into spherical nanoparticles [42]. | Effectively protects dsRNA from RNase A degradation; improves leaf deposition and adhesion [42]. | Improved RNAi efficiency and prolonged protective duration against fungal pathogen Rhizoctonia solani [42]. |
Competitive Inhibition with dsDNA
A novel biochemical approach involves using double-stranded DNA (dsDNA) as a competitive substrate for non-specific nucleases (NSEs). In the brown marmorated stink bug (Halyomorpha halys), dsRNA is rapidly degraded in saliva by HhNSE. Co-formulating dsRNA with dsDNA competitively inhibits HhNSE, protecting the dsRNA from degradation and significantly enhancing target gene silencing in vivo [43].
Detailed Experimental Protocols
Protocol: ZIF-8@PDA Nanocarrier for dsRNA Delivery
This protocol is adapted from methods used to enhance RNAi in lepidopteran pests [41].
Reagents:
- Zinc nitrate hexahydrate (Zn(NO₃)₂·6H₂O)
- 2-Methylimidazole (2-mIm)
- Dopamine hydrochloride
- Tris-HCl buffer (10 mM, pH 8.5)
- Nuclease-free dsRNA solution (0.1-0.5 µg/µL)
Procedure:
- Synthesis of ZIF-8 Core: Dissolve 2.93 g of Zn(NO₃)₂·6H₂O and 3.24 g of 2-mIm separately in 100 mL of methanol each. Rapidly mix the two solutions and stir for 1 hour at room temperature. Recover the white precipitate by centrifugation (10,000 × g, 15 min), wash three times with methanol, and dry under vacuum.
- Loading of dsRNA: Dissolve 10 mg of ZIF-8 in 1 mL of nuclease-free water. Add 1 mL of dsRNA solution (e.g., 200 µg total) and incubate for 30 minutes with gentle agitation.
- Polydopamine Coating: Add 10 mg of dopamine hydrochloride to the dsRNA@ZIF-8 suspension. Adjust the pH to 8.5 using Tris-HCl buffer and stir for 6 hours at room temperature. The solution will darken.
- Purification: Recover the resulting dsRNA@ZIF-8@PDA nanoparticles by centrifugation (12,000 × g, 20 min), wash twice with nuclease-free water, and re-suspend in water or buffer for application.
- Characterization: Determine particle size and zeta potential using Dynamic Light Scattering (DLS). Confirm morphology and size distribution using Scanning Electron Microscopy (SEM) or Transmission Electron Microscopy (TEM).
Protocol: Competitive Inhibition Assay Using dsDNA
This protocol describes an ex vivo assay to test the protective effect of dsDNA on dsRNA, based on research in Halyomorpha halys [43].
Reagents:
- Insect saliva or gut fluid (collected and pooled from target species)
- Target dsRNA (e.g., 500 ng/µL)
- Competitor dsDNA (e.g., PCR-amplified fragment or salmon sperm DNA)
- Reaction buffer (e.g., 10 mM Tris-HCl, pH 7.5)
- Gel loading dye and agarose for electrophoresis
Procedure:
- Prepare Reaction Mixtures:
- Test: 2 µL dsRNA + 2 µL dsDNA (at various mass ratios, e.g., 1:1, 1:2) + 6 µL saliva/gut fluid.
- Degradation Control: 2 µL dsRNA + 6 µL saliva/gut fluid + 2 µL nuclease-free water.
- dsRNA Integrity Control: 2 µL dsRNA + 8 µL nuclease-free water.
- Incubate: Place all reaction mixtures at the insect's physiological temperature (e.g., 25-28°C) for a time-course (e.g., 0, 1, 5, 10, 30 minutes).
- Stop Reaction: Heat-inactivate the samples at 75°C for 10 minutes after each time point, or add an equal volume of gel loading dye containing EDTA.
- Analyze Integrity: Load the reactions on a 1% agarose gel. A clear, intact dsRNA band in the test mixture compared to the degraded control indicates successful protection by dsDNA.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for dsRNA Stability Research
| Reagent / Material | Function in Research | Specific Example |
|---|---|---|
| Aluminum Sulfate | Chemical co-treatment to remove persistent PCR inhibitors from complex matrices like soil [44]. | Used in optimized soil dsRNA extraction to recover ~80% of spiked dsRNA [44]. |
| β-Mercaptoethanol (β-ME) | A reducing agent used to inhibit RNases during nucleic acid extraction [44]. | Component of optimized lysis buffer for dsRNA extraction from clay and sandy soils [44]. |
| Polyvinylpyrrolidone (PVP) | Adsorbs phenolic compounds and other enzymatic inhibitors commonly found in biological samples [44]. | Used in soil dsRNA extraction protocols to improve purity and downstream qRT-PCR compatibility [44]. |
| TRI Reagent | A monophasic solution of phenol and guanidine isothiocyanate for simultaneous liquid-phase separation of RNA, DNA, and proteins [44]. | Base reagent for standard nucleic acid extraction; requires optimization for dsRNA recovery from soil [44]. |
| HT115 (DE3) E. coli Strain | An RNase III-deficient engineered bacterial strain for high-yield, low-cost production of dsRNA [41]. | Cost-effective synthesis of dsRNA for large-scale bioassays and field applications [41]. |
RNA interference (RNAi) presents a promising strategy for controlling insect pests by silencing genes essential for fecundity and egg hatchability [45]. A significant challenge in this field is the efficient cellular uptake of double-stranded RNA (dsRNA), the effector molecule in RNAi pathways. This application note details protocols and mechanistic insights for enhancing dsRNA delivery into insect cells by leveraging systemic RNA interference-deficient (SID)-1 homologues and advanced nanomaterial carriers, with specific application to research aimed at reducing insect fertility.
Table 1: Key Proteins in dsRNA Uptake and Their Functions
| Protein Name | Organism | Function in dsRNA Uptake | Cellular Localization |
|---|---|---|---|
| SID-1 [46] | C. elegans | Putative dsRNA channel; mediates systemic RNAi | Cell membrane |
| SIDT2 [47] | Mammals | Nucleic acid transporter; mediates dsRNA transport into cytoplasm | Lysosomal/Endosomal membrane |
| LmSRA, LmSRC [48] | L. migratoria | Scavenger receptors; bind dsRNA-carrier complexes for endocytosis | Cell membrane |
| LmLPR, LmLRP1-3 [48] | L. migratoria | Lipoprotein receptors; bind dsRNA-carrier complexes for endocytosis | Cell membrane |
| LmV-ATPase [48] | L. migratoria | Proton pump; acidifies endosomes to facilitate dsRNA escape | Endosomal membrane |
Mechanisms of dsRNA Cellular Import
SID-1 Homologues and Transmembrane Transport
The SID-1 family of transmembrane proteins facilitates the systemic spread of RNAi. Recent structural studies reveal that SID-1 proteins function as dsRNA-gated channels [46]. Cryo-EM structures show that SID-1 specifically recognizes dsRNA in a sequence-independent manner through extensive ionic interactions between basic residues and the phosphate backbone, as well as hydrogen bonds with the 2'-hydroxyl group of the RNA [49]. This mechanism allows SID-1 to distinguish between dsRNA and dsDNA [49]. In the context of insect pest control, enhancing the native function of SID-1 homologues could significantly improve systemic RNAi efficiency, a key factor in achieving robust silencing of fertility-related genes.
Endocytic Pathways and Intracellular Trafficking
In insects where SID-1-like proteins are absent or less effective, dsRNA relies on receptor-mediated endocytosis for cellular entry. In the fat body of Locusta migratoria, a key tissue for metabolic and reproductive functions, the pathway involves:
- Carrier Binding: dsRNA in the hemolymph is bound by carrier proteins like apolipophorin-III (ApoLp-III) [48].
- Receptor Recognition: The dsRNA-ApoLp-III complex is recognized by membrane receptors, including scavenger receptors (LmSRA, LmSRC) and lipoprotein receptors (LmLPR, LmLRP1-3) [48].
- Cellular Internalization: The complex is internalized via clathrin-mediated endocytosis and macropinocytosis [48].
- Intracellular Transport and Escape: Internalized vesicles are trafficked through early and late endosomes, a process guided by Rab GTPases (LmRab4, LmRab7, LmRab9). The vacuolar-type H+-ATPase (V-ATPase) acidifies the endosome, facilitating the escape of dsRNA into the cytoplasm where it can engage the RNAi machinery [48].
Figure 1: Pathway of Receptor-Mediated dsRNA Uptake and Intracellular Trafficking in Insect Cells. This diagram illustrates the key steps from extracellular dsRNA binding to its eventual release into the cytoplasm, a critical process for successful gene silencing in fertility research.
Advanced Carrier Systems for Enhanced Delivery
Nanomaterial-based carriers protect dsRNA from degradation and enhance cellular uptake.
Table 2: Nanomaterial Carriers for dsRNA Delivery
| Carrier Type | Composition | Mechanism of Uptake | Reported Efficacy | Application Context |
|---|---|---|---|---|
| Lipid Nanoparticles (LNPs) [50] | Ionizable lipids, phospholipids, cholesterol, PEG-lipids | Endocytosis | Used in clinical trials for siRNA delivery in humans [50] | Human therapeutics |
| Mesoporous Silica Nanoparticles (MSNs) [51] | Silica-based porous structures | Clathrin-mediated endocytosis | Significant reduction in clubroot disease severity in rapeseed [51] | Plant disease control |
| Cationic Polymers [50] | Cyclodextrin-containing polymers, polyethylenimine (PEI) | Endocytosis | Used in clinical-stage RNAi cancer therapy (CALAA-01) [50] | Human therapeutics |
Experimental Protocols
Protocol: Assessing the Role of SID-1 Homologues in dsRNA Uptake
Objective: To evaluate the functional role of SID-1 homologues in dsRNA uptake and its impact on silencing fertility-related genes.
Materials:
- Cultured insect cell lines (e.g., Drosophila S2, lepidopteran, or coleopteran cells).
- dsRNA targeting a gene of interest (e.g., a vitellogenin receptor for fecundity studies) and a control (e.g., GFP).
- Fluorescently labeled dsRNA (e.g., Cy3-dsRNA).
- siRNA targeting the SID-1 homologue transcript.
- Transfection reagent.
- Confocal microscope.
- qRT-PCR equipment.
- Flow cytometer.
Procedure:
- Gene Knockdown: Transfect cells with siRNA targeting the SID-1 homologue or a non-targeting control siRNA.
- Incubation: Incubate cells for 48-72 hours to allow for protein knockdown.
- dsRNA Uptake Assay:
- Treat siRNA-pre-treated cells with fluorescently labeled dsRNA.
- Incubate for 2-4 hours.
- Wash cells thoroughly to remove extracellular dsRNA.
- Analyze intracellular fluorescence using flow cytometry (quantitative) or confocal microscopy (for visual confirmation and localization).
- Functional Validation:
- In parallel, treat cells with dsRNA targeting a fertility gene.
- After 48-96 hours, harvest cells and extract total RNA.
- Perform qRT-PCR to quantify the mRNA levels of the target fertility gene.
- Correlate the knockdown efficiency with the level of SID-1 homologue expression.
Protocol: Evaluating Nanocarrier-Mediated dsRNA Delivery
Objective: To test the efficacy of mesoporous silica nanoparticles (MSNs) in delivering dsRNA and silencing target genes in insect tissues.
Materials:
- Synthesized and characterized MSNs.
- Target dsRNA.
- Fluorescent dye (e.g., FITC) for labeling MSNs.
- Insect diet or artificial feeding system.
- Dissection tools.
- Confocal microscope.
- qRT-PCR equipment.
Procedure:
- Complex Formation: Load dsRNA onto MSNs by mixing in nuclease-free water or buffer. Incubate with shaking for 2 hours at room temperature.
- Delivery:
- Option A (Oral Delivery): Mix the dsRNA-MSN complex with an artificial diet and feed it to adult insects. A control group should receive diet with naked dsRNA.
- Option B (Topical Application/Tissue Injection): For precise dosing, inject the complex directly into the hemolymph or apply it topically.
- Uptake and Localization Analysis:
- After 24 hours, dissect target tissues (e.g., fat body, ovaries).
- For fluorescently labeled MSNs, image tissues using a confocal microscope to confirm uptake and intracellular localization.
- Gene Silencing Assessment:
- After 3-5 days, dissect tissues and extract total RNA.
- Perform qRT-PCR to measure the transcript levels of the target fertility gene.
- Monitor downstream phenotypic effects such as egg-laying rate (fecundity) and egg hatchability over the subsequent days or weeks.
Figure 2: Workflow for Evaluating Nanocarrier-Mediated dsRNA Delivery. This protocol outlines the key steps from preparing the dsRNA-nanocarrier complex to final assessment of gene silencing and phenotypic effects on fertility.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for dsRNA Uptake and Fertility Research
| Reagent / Material | Function/Description | Example Application |
|---|---|---|
| SID-1/sid-1 Antibodies [46] [49] | Detect and localize SID-1 homologue protein expression in insect tissues. | Validate protein expression in ovaries and fat body via immunohistochemistry. |
| Fluorescently-Labeled dsRNA (e.g., Cy3-dsRNA) [48] | Visualize and quantify the uptake and trafficking of dsRNA in cells and tissues. | Track dsRNA internalization in live or fixed tissues using confocal microscopy. |
| Apolipophorin-III (ApoLp-III) [48] | Recombinant insect carrier protein that binds dsRNA and facilitates recognition by membrane receptors. | Pre-complex with dsRNA to enhance stability and uptake in hemolymph-feeding assays. |
| V-ATPase Inhibitors (e.g., Bafilomycin A1) [48] | Block endosomal acidification, preventing dsRNA escape and allowing study of this critical step. | Investigate the role of endosomal escape in the overall RNAi efficiency pathway. |
| Mesoporous Silica Nanoparticles (MSNs) [51] | Nanocarrier that protects dsRNA and promotes cellular uptake via endocytosis. | Formulate with dsRNA for oral delivery to test protection from gut nucleases. |
| Clathrin-Mediated Endocytosis Inhibitors (e.g., Pitstop 2) [48] | Specifically inhibit clathrin-dependent uptake, allowing dissection of entry pathways. | Determine the primary mechanism of dsRNA entry in a given cell type (e.g., ovarian). |
| Rab GTPase siRNAs [48] | Knockdown specific Rab proteins (e.g., Rab7) to disrupt intracellular vesicular trafficking. | Elucidate the role of specific trafficking steps in successful gene silencing. |
In RNA interference (RNAi) research aimed at reducing fecundity and egg hatchability, the precise design of double-stranded RNA (dsRNA) is paramount for achieving effective gene silencing while minimizing off-target effects. Off-target effects occur when dsRNA inadvertently silences genes with partial sequence complementarity, potentially compromising experimental validity and raising safety concerns for therapeutic applications. The core of this challenge lies in the RNAi mechanism itself: after cellular uptake, dsRNA is processed by the Dicer enzyme into small interfering RNAs (siRNAs) of 20-23 nucleotides. These siRNAs are then loaded into the RNA-induced silencing complex (RISC), which uses the guide (antisense) strand to find and cleave complementary messenger RNA (mRNA) targets [52] [53]. Off-target silencing primarily happens when the "seed region" (nucleotides 2-8 of the siRNA guide strand) has sufficient complementarity to non-target mRNAs, leading to their unintended degradation or translational repression [54].
Advancements in bioinformatics have identified key sequence and structural features that influence both the efficacy and specificity of dsRNA. Research in the red flour beetle, Tribolium castaneum, has revealed that thermodynamic asymmetry in the siRNA duplex is a critical predictive feature for high efficacy and reduced off-target effects. The strand with the less tightly paired 5' end is preferentially selected by RISC as the guide strand. Biasing this selection toward the antisense strand ensures accurate targeting and reduces the chance of the sense strand causing off-target effects [52]. Furthermore, the nucleotide composition, particularly a high GC content between the 9th and 14th nucleotides of the antisense siRNA, is associated with increased efficacy in insects, a finding that contrasts with data from human cells [52]. The presence of an adenine at the 10th position in the antisense strand has also been correlated with high insecticidal efficacy [52]. Finally, minimizing the formation of intramolecular secondary structures within the dsRNA sequence ensures better processing by Dicer and availability of the siRNA guide strand [52] [55]. These principles form the foundation for rational dsRNA design, which can be operationalized using specialized bioinformatics tools.
Bioinformatics Tools for Rational dsRNA Design
To translate design principles into practice, researchers can leverage several web-based platforms that automate the selection of optimal dsRNA sequences. These tools perform comprehensive analyses against entire transcriptomes to maximize on-target efficiency while minimizing risks to non-target organisms. The table below summarizes two leading tools for this purpose.
Table 1: Comparison of Bioinformatics Tools for dsRNA Design
| Tool Name | Key Functionalities | Notable Features | Application Context |
|---|---|---|---|
| dsRIP [52] | - Optimizes dsRNA sequences for efficacy- Identifies effective target genes- Minimizes risk to non-target species | Incorporates insect-specific siRNA efficacy parameters (e.g., thermodynamic asymmetry, GC content in specific regions). | Pest control research; designing species-specific dsRNA for laboratory and field applications. |
| dsRNAEngineer [56] | - Screen-target analysis- On-target analysis- Off-target analysis- Multi-target analysis | Hosts 941 transcriptomes for comprehensive on-/off-target assessment; enables design of dsRNAs that target multiple pest species simultaneously (cotargeting). | Ecological risk assessment; designing dsRNAs for multi-pest control while protecting beneficial species. |
These platforms address the critical need for on-target efficacy—ensuring the dsRNA effectively silences the intended gene in the pest or research organism—and off-target safety—preventing the silencing of genes in non-target species, such as beneficial insects, predators, and pollinators [56]. The "screen-target" function in dsRNAEngineer is particularly useful for fecundity research, as it can identify conserved genes suitable for cotargeting across multiple related pest species [56].
Quantitative Parameters for Effective siRNA Design
The ultimate efficacy and specificity of a long dsRNA molecule depend on the collective properties of the siRNAs processed from it. Since insects lack the robust secondary siRNA amplification machinery found in C. elegans, they rely heavily on the primary siRNA pool derived directly from the delivered dsRNA, making the parent dsRNA sequence critically important [52]. Systematic screening of individual siRNA efficacy has yielded quantitative parameters that reliably predict performance.
Table 2: Key Sequence Features for Optimizing siRNA Efficacy and Specificity
| Feature | Description | Impact on Efficacy & Specificity |
|---|---|---|
| Thermodynamic Asymmetry | Difference in binding stability at the 5' ends of the two siRNA strands. | Promotes correct RISC loading of the antisense guide strand, reducing off-target effects mediated by the sense strand [52]. |
| GC Content (nt 9-14) | Proportion of Guanine and Cytosine bases in the central region of the antisense siRNA. | High GC content in this region is predictive of high efficacy in insects [52]. |
| Nucleotide at Position 10 | The base at the 10th position of the antisense siRNA strand. | Adenine (A) at this position is strongly associated with high efficacy [52]. |
| Secondary Structure | Intramolecular base-pairing within the dsRNA or siRNA. | The absence of stable secondary structures in the target mRNA region and the dsRNA itself predicts higher efficacy [52] [55]. |
| siRMSD (siRNA Root-Mean-Square Deviation) | A parameter that quantifies structural distortion caused by chemical modifications [54]. | Higher siRMSD values correlate with reduced off-target effects by disrupting canonical A-form RNA geometry and seed region interactions [54]. |
These parameters provide a blueprint for designing highly effective and specific dsRNA. For instance, selecting a target region within an mRNA that, when processed, yields siRNAs rich in these features will significantly improve the RNAi outcome. The parameter siRMSD is especially relevant for advanced therapeutic applications, as it helps rationalize the impact of chemical modifications introduced to improve siRNA stability and reduce immunogenicity [54].
Experimental Protocol: RNAi in Insect Eggs via Egg-Soaking
Targeting the egg stage is a strategic approach for research focused on reducing fecundity and hatchability. The following protocol, adapted from successful experiments on Spodoptera littoralis and Sarcoptes scabiei, details a robust method for inducing RNAi in insect eggs via dsRNA soaking [57] [1].
Materials and Reagents
Table 3: Essential Research Reagent Solutions for RNAi Egg-Soaking
| Reagent / Material | Function / Purpose | Example Specification / Notes |
|---|---|---|
| Gene-Specific dsRNA | The active silencing molecule. | 200-500 bp fragment from target gene; concentration 50-250 ng/µL for soaking [57] [1]. |
| Control dsRNA | Control for non-sequence-specific effects. | dsRNA targeting a non-endogenous gene (e.g., GFP) [1]. |
| Sodium Hypochlorite (NaOCl) | Permeabilizing agent for the eggshell. | 2% solution; pre-treatment time is critical and must be optimized [57]. |
| Phosphate-Buffered Saline (PBS) | Physiological buffer for dsRNA dilution and soaking. | 1X concentration, sterile [1]. |
| TRIzol Reagent | For total RNA extraction from eggs/larvae for validation. | - |
| qRT-PCR System | Quantitative assessment of target gene knockdown. | Requires gene-specific primers and a suitable reference gene [57] [1]. |
Step-by-Step Procedure
- dsRNA Preparation: Synthesize and purify dsRNA corresponding to the target gene essential for embryogenesis or egg development. A common method is in vitro transcription using T7 RNA polymerase. Resuspend the final dsRNA pellet in nuclease-free 1X PBS to a working concentration of 50-250 ng/µL, or higher (e.g., 2.5 µg/µL) if pre-treatment is used [57] [1]. Aliquot and store at -80°C.
- Egg Collection and Preparation: Collect freshly laid egg masses (within 30 minutes of oviposition) and gently separate individual eggs using a fine brush to ensure highly synchronized embryonic development [1].
- Eggshell Pre-treatment (Optional but Recommended): For species with robust egg chorions that impede dsRNA uptake, pre-treat eggs with a permeabilization agent. Immerse approximately 120 eggs in a 2% sodium hypochlorite (NaOCl) solution for a brief, empirically determined period. Immediately after treatment, wash the eggs thoroughly with physiological saline (e.g., PBS) to remove the NaOCl [57].
- dsRNA Soaking: Transfer the pre-treated (or untreated) eggs to a 1.5 mL microcentrifuge tube. Soak them in 50-100 µL of the dsRNA solution. The optimal incubation conditions (temperature and duration) vary by species:
- Post-Soaking Incubation and Phenotypic Assessment: After soaking, carefully transfer the eggs to a fresh Petri dish with a suitable substrate. Incubate them at the optimal temperature for embryonic development (e.g., 37°C for S. scabiei [57]) under high-humidity conditions. Monitor the eggs daily to record the hatching rate. Observe and document any phenotypic alterations in the embryos or hatched larvae, such as developmental delays or morphological abnormalities [57] [1].
- Validation of Gene Silencing (qRT-PCR): To confirm the RNAi effect at the molecular level, extract total RNA from a subset of treated eggs (or hatched larvae) at a relevant developmental stage using TRIzol reagent. Perform absolute quantitative Real-Time PCR (qRT-PCR) to measure the transcript levels of the target gene relative to a stable reference gene. A successful knockdown should show a significant reduction (e.g., 65-85%) in target mRNA levels compared to the control group [57] [1].
This protocol provides a foundational framework that can be adapted and optimized for specific insect species and research goals in fecundity and hatchability studies.
Within the broader research on using RNA interference (RNAi) to reduce insect fecundity and egg hatchability, a central challenge persists: the variable silencing efficiency observed across different experiments. Achieving consistent and potent gene silencing depends on a triad of critical, interdependent factors: the selection of effective target genes, the determination of an optimal double-stranded RNA (dsRNA) dosage, and the careful planning of exposure duration. This Application Note synthesizes recent research to provide a structured framework for optimizing these parameters, with a specific focus on applications aimed at impairing reproductive success in insect pests. The protocols and data summarized herein are designed to equip researchers with practical strategies to enhance the efficacy and reproducibility of their RNAi-based experiments.
The following tables consolidate key quantitative findings from recent studies, highlighting the impact of target gene selection, dsRNA dosage, and exposure time on RNAi efficacy related to fecundity and egg hatchability.
Table 1: Impact of Target Gene Selection and dsRNA Dosage on Reproductive Performance
| Target Insect Species | Target Gene | dsRNA Dosage | Exposure Duration | Key Efficacy Outcomes (Fecundity & Hatchability) | Citation |
|---|---|---|---|---|---|
| Aethina tumida (Small Hive Beetle) | JHAMT | Oral feeding (dsRNA-SPc mix) | Not Specified | Reduced female fecundity, fertility, and egg hatchability; rescued by methoprene application. [16] | |
| Laodelphax striatellus (Small Brown Planthopper) | LsTH / LsDDC | Ingestion or injection | Not Specified | Shortened oviposition period, reduced fecundity, inhibited egg hatchability and development. [10] | |
| Spodoptera littoralis (Cotton Leafworm) | Sl102 | Egg soaking (250 ng/µL) | 120 minutes | Drastic reduction in egg hatching rate; high mortality of hatched larvae. [1] | |
| Agrilus planipennis (Emerald Ash Borer) | hsp / shi | 1 µg/µL (larva); 10 µg/µL (adult) | 8 days (larvae) | Up to 93.3% larval mortality with dsHSP; 90% adult mortality with dsHSP+dsSHI mix. [58] | |
| Tuta absoluta (Tomato Pinworm) | CYP9A306 / CYB5R | Nanocarrier (SPc)-mediated delivery | Not Specified | Increased susceptibility to insecticide; fitness costs including reduced fecundity and hatching rate. [59] |
Table 2: Efficacy of Target Genes in Embryonic and Larval Stages
| Target Gene | Biological Function | Demonstrated Efficacy in Embryos | Demonstrated Efficacy in Larvae/Adults | Key Phenotypic Outcomes | Citation |
|---|---|---|---|---|---|
| JHAMT | Juvenile hormone synthesis | Not directly tested | High | Depressed ovarian development, reduced fecundity and egg hatchability. [16] | |
| TH / DDC | Dopamine synthesis | High (via parental RNAi) | High | Impaired reproduction, reduced vitellogenin expression, inhibited egg hatching. [10] | |
| Sl102 | Amyloid fibrils for basal lamina formation | High | High (immune suppression) | Disrupted embryonic development, drastic reduction in egg hatching. [1] | |
| hsp / shi | Stress response / Endocytosis | Not tested | Very High | High mortality in both larvae and adults. [58] |
Experimental Protocols
This section outlines detailed methodologies for key experiments cited in this note, providing reproducible protocols for researchers.
Protocol: RNAi via Egg Soaking for Embryonic Suppression
- Application: Targeted suppression of embryonic development to reduce egg hatchability. [1]
- Materials:
- Highly synchronized insect eggs (laid within a 30-min window).
- Target-specific dsRNA (e.g., dsSl102) and control dsRNA (e.g., dsGFP).
- Phosphate-Buffered Saline (PBS), pH 7.4.
- 1.5 mL Eppendorf tubes.
- Procedure:
- Egg Collection: Collect approximately 120 synchronized eggs into a 1.5 mL tube.
- dsRNA Solution Preparation: Prepare a solution of dsRNA in PBS. A concentration of 250 ng/µL is recommended based on efficacy studies. [1]
- Soaking: Add 50 µL of the dsRNA solution to the tube containing eggs, ensuring full immersion.
- Incubation: Soak the eggs for 120 minutes at 25 ± 1 °C.
- Post-treatment: After soaking, remove the solution and transfer the eggs to appropriate conditions for development.
- Assessment: Monitor and record the egg hatching rate and observe morphological alterations in the embryos.
Protocol: Oral Delivery of dsRNA for Larval/Adult RNAi
- Application: Silencing genes in larval or adult stages to assess impacts on fecundity, fertility, and survival. [16] [58]
- Materials:
- Procedure:
- dsRNA Complexation: For enhanced stability and uptake, complex dsRNA with SPc nanoparticles by mixing and incubating at room temperature for 20-30 minutes. [16] [59]
- Delivery Mixture Preparation: Incorporate the dsRNA-SPc complex into an artificial diet (for larvae) or a sucrose solution (for adults).
- Feeding: Present the treated diet or solution to the insects. For neonates, a feeding period of 8-10 days is common, with mortality assessed periodically. [58]
- Rescue Experiments: To confirm target specificity, a rescue experiment can be performed by simultaneously applying methoprene (a JH analog) when targeting the JH pathway. [16]
- Evaluation: Record mortality, oviposition period, fecundity, and egg hatchability. Confirm gene knockdown via qRT-PCR.
Signaling Pathways in Reproductive RNAi
The following diagrams illustrate the logical relationships and signaling pathways through which RNAi targeting key genes leads to reduced fecundity and egg hatchability.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Materials for RNAi Fecundity Research
| Reagent / Material | Function & Application in RNAi Research | Key Considerations |
|---|---|---|
| Star Polycation (SPc) | A nanocarrier that spontaneously binds to dsRNA via electrostatic interactions, shielding it from degradation and enhancing cellular uptake. Crucial for oral delivery in many insects. [16] [59] | Improves dsRNA stability against nucleases; enhances silencing efficacy, especially in lepidopterans and other insects with robust RNAi degradation systems. |
| dsRNA Targeting Reproductive Genes | The core effector molecule for gene silencing. Targets such as JHAMT, TH, DDC, and Vg are critical for investigating fecundity and hatchability. [16] [10] | Requires high-quality, nuclease-free synthesis. Sequence specificity must be verified to minimize off-target effects. |
| Methoprene (JH Analog) | Used in rescue experiments to confirm the specificity of RNAi targeting the juvenile hormone pathway. Application should partially restore reproductive parameters silenced by dsJHAMT. [16] | Serves as a critical control for validating that the observed phenotype is due to specific pathway disruption. |
| Nuclease-Free Buffers (e.g., PBS) | Used as a solvent for dsRNA in egg soaking and other delivery methods. Essential for maintaining RNA integrity during experimental procedures. [1] | Prevents dsRNA degradation before cellular uptake. Critical for reproducibility in soaking and feeding assays. |
| Chemically Modified siRNA | siRNA with modifications (e.g., 2'-O-methyl) to the ribose backbone enhance stability and prolong silencing duration, which is vital for therapeutic development. [60] [61] | Modification patterns must be optimized as they can significantly impact efficacy and potentially increase off-target risks. |
Proof of Concept: Validating Efficacy and Comparing RNAi to Alternative Technologies
Within the broader thesis on using RNA interference (RNAi) to control pest populations, the phenotypic assessment of reduced fecundity and egg hatchability serves as a critical measure of intervention success. RNAi technology functions by silencing essential genes involved in reproduction, leading to compromised ovarian development, reduced egg-laying capacity, and impaired embryonic development [62]. This application note provides detailed protocols and quantitative frameworks for assessing these key phenotypic outcomes, enabling researchers to accurately evaluate the efficacy of RNAi-based strategies in reducing insect fertility.
The core mechanism involves introducing sequence-specific double-stranded RNA (dsRNA) that degrades complementary messenger RNA (mRNA) transcripts of target genes, thus preventing the synthesis of proteins vital for reproduction [63]. Genes such as vitellogenin (Vg) and its receptor (VgR) have been identified as promising targets, as they play indispensable roles in yolk protein uptake and oocyte maturation [62]. This document standardizes the methodologies for quantifying the resulting phenotypic effects, which is essential for validating gene targets and optimizing dsRNA delivery systems, such as nanoclay carriers [64] and transgenic plants [65].
Key Genetic Targets and Observed Phenotypic Effects
Silencing specific genes involved in insect reproduction leads to measurable declines in fertility. The table below summarizes high-value targets and the resulting phenotypic outcomes from recent studies.
Table 1: Key Genetic Targets for RNAi-Mediated Reduction of Fecundity and Hatch Rates
| Target Gene | Insect Species | Fecundity Reduction | Hatch Rate Reduction | Other Phenotypes |
|---|---|---|---|---|
| Vitellogenin (LsVg) | Lasioderma serricorne (Cigarette Beetle) | Significant decrease in number of eggs laid [62] | Significant decrease in egg hatchability [62] | Impaired ovarian development; decreased oocyte length [62] |
| Vitellogenin Receptor (LsVgR) | Lasioderma serricorne (Cigarette Beetle) | Significant decrease in number of eggs laid [62] | Significant decrease in egg hatchability [62] | Impaired ovarian development; decreased oocyte length [62] |
| Ryanodine Receptor (BtRyR) | Bemisia tabaci (Whitefly) | Reduced egg laying (48.19% to 10.81% of control) [64] | Delayed adult emergence (34.88% to 7.26% of control) [64] | Increased mortality (60–100%) [64] |
| nAChR-β1 (BtnAChR-β1) | Bemisia tabaci (Whitefly) | Reduced egg laying (48.19% to 10.81% of control) [64] | Delayed adult emergence (34.88% to 7.26% of control) [64] | Increased mortality (60–100%) [64] |
| Trehalose-6-Phosphate Synthase (BtTPS1/BtTPS2) | Bemisia tabaci (Whitefly) | Decreased fecundity in adults [65] | Low hatchability in nymphs; 90% mortality [65] | Retarded growth in nymphs [65] |
RNAi Mechanisms and Experimental Workflow
The following diagram illustrates the core mechanism of RNAi and its impact on insect reproduction, followed by a generalized experimental workflow for conducting and evaluating an RNAi fecundity study.
Detailed Experimental Protocols
Protocol A: dsRNA Preparation and Microinjection in Coleoptera
This protocol is adapted from studies on Lasioderma serricorne and is suitable for other beetle species [62].
- dsRNA Synthesis: Design primers with T7 promoter sequences for the target gene (e.g., LsVg or LsVgR). Use a transcription kit to synthesize dsRNA. Resuspend the final dsRNA pellet in nuclease-free water or buffer to a recommended stock concentration of 5 mg/mL [66].
- Insect Microinjection: Anesthetize adult female insects (e.g., 1-2 days post-eclosion) on ice. Using a microinjector, deliver a defined volume of dsRNA (e.g., 200-500 nL) into the insect's hemocoel, typically in the thoracic or abdominal region. Control groups should be injected with a similar volume of dsRNA targeting a non-insect gene (e.g., GFP). A minimum of 30-50 individuals per treatment group is recommended for robust statistical analysis [62].
- Post-Injection Rearing: Maintain injected insects under standard conditions (e.g., 28°C ± 1°C, 40% ± 5% relative humidity). Provide a suitable diet. Monitor mortality daily.
Protocol B: Foliar and Root Application via Nanocarriers in Hemiptera
This protocol utilizes clay nanosheets as carriers for dsRNA delivery, as demonstrated in whitefly management [64].
- dsRNA-Nanoclay Complex Preparation: Synthesize clay nanosheets via the hydrothermal method. Complex dsRNA with the nanosheets at a typical loading ratio of 1:10 (dsRNA:Nanoclay). Prepare the complex in various concentrations for dose-response assays (e.g., 20, 40, and 60 µg/mL) [64].
- Plant Application:
- Foliar Spray: Apply the dsRNA-nanoclay solution evenly to the abaxial and adaxial surfaces of plant leaves using a hand-held sprayer until runoff.
- Root Dip: Uproot seedlings, gently wash the roots, and immerse them in the dsRNA-nanoclay solution for a specified period before replanting.
- Insect Bioassay: After application, introduce adult insects to the treated plants. Encase the plant in a vented cage to contain the insects. The root dip method has been shown to have higher silencing efficiency than foliar spray [64].
Protocol C: Phenotypic Assessment and Data Collection
This is a universal protocol for quantifying the key phenotypic outcomes following an RNAi treatment.
- Fecundity Assay: Following treatment, house individual female insects or defined groups and provide an oviposition substrate. Count the number of eggs laid daily over the entire oviposition period. Record the oviposition period and the total number of eggs per female [62].
- Hatchability Assay: After the egg-laying period, monitor the eggs daily. The number of hatched larvae should be recorded. Calculate the egg hatch rate for each replicate using the formula: (Number of Hatched Eggs / Total Number of Eggs Laid) × 100% [62].
- Morphological Assessment: Dissect female insects in phosphate-buffered saline (PBS) under a microscope. Examine the ovaries and measure the length of ovarian tubes and oocytes using calibrated ocular micrometer. Compare these measurements to control groups to assess developmental impairment [62].
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Key Research Reagent Solutions for RNAi Fecundity Studies
| Item | Function/Description | Example Usage |
|---|---|---|
| In Vivo Ready siRNA/dsRNA | Chemically synthesized RNA duplexes, formulated for stability and minimal immune response in living organisms. | Resuspended in nuclease-free buffer for microinjection [66]. |
| T7 or U6 Promoter Plasmids | Vectors for in vitro transcription of dsRNA or for cellular expression of short-hairpin RNA (shRNA). | Template for dsRNA synthesis targeting insect genes [62]. |
| Nanoclay Carriers (e.g., LDH) | Layered double hydroxide particles that bind and protect dsRNA, facilitating delivery via feeding or spraying. | Complex with dsRNA for foliar application against whiteflies [64]. |
| Microinjection System | Comprises a micromanipulator, microinjector, and capillary needles for precise delivery of dsRNA into insects. | Injection of dsRNA into the hemocoel of adult beetles [62]. |
| TRIzol Reagent | A ready-to-use monophasic solution for the isolation of high-quality total RNA from cells and tissues. | RNA extraction from insect ovaries to confirm gene knockdown via qPCR [66]. |
| SYBR Green qPCR SuperMix | A master mix for quantitative real-time PCR (qRT-PCR) to measure gene expression knockdown. | Validation of target gene silencing (e.g., LsVg) post-RNAi treatment [62]. |
Data Analysis and Validation
Robust statistical analysis is paramount. Compare fecundity and hatch rates between treatment and control groups using appropriate tests, such as Student's t-test for two groups or ANOVA followed by post-hoc tests for multiple groups. A p-value of less than 0.05 is typically considered statistically significant. The data should be presented as mean ± standard error.
- Gene Knockdown Validation: Use quantitative real-time PCR (qRT-PCR) to confirm the reduction in target mRNA levels. The 2−∆∆CT method is commonly used to calculate relative expression changes, with stable reference genes (e.g., EF1a and 18S) for normalization [62].
- Off-Target Effect Controls: To ensure phenotypic effects are due to specific gene silencing, include control groups injected with non-targeting dsRNA (e.g., GF). Furthermore, use BLAST analysis to ensure the dsRNA sequence is unique to the target gene, minimizing the risk of off-target silencing via partial complementarity, particularly in the seed region (nucleotides 2-7) of the siRNA guide strand [67].
RNA interference (RNAi) has revolutionized functional genomics and therapeutic development by enabling sequence-specific gene silencing. Within research aimed at reducing fecundity and egg hatchability—a promising strategy for pest control and reproductive health applications—accurate confirmation of silencing is paramount. Molecular validation through quantitative reverse transcription PCR (qRT-PCR) and transcriptomics provides essential evidence of target gene knockdown and investigation of potential off-target effects. This application note details standardized protocols and critical considerations for confirming RNAi efficacy, drawing from recent advances in methodology and instrumentation to ensure reliable, reproducible results for researchers and drug development professionals.
RNAi Mechanism and Validation Principles
RNAi functions through the introduction of double-stranded RNA (dsRNA), which is processed by the Dicer enzyme into small interfering RNAs (siRNAs) of 21-25 nucleotides [68]. These siRNAs are incorporated into the RNA-induced silencing complex (RISC), guiding it to complementary mRNA sequences for cleavage and degradation [68]. This process results in post-transcriptional gene silencing (PTGS), reducing the abundance of the target transcript and its corresponding protein.
The core principle of molecular validation is to quantitatively measure this reduction in target mRNA levels following RNAi treatment. While western blotting confirms knockdown at the protein level, qRT-PCR provides a more sensitive and rapid assessment of mRNA silencing. Transcriptomics, through microarray analysis or RNA sequencing, offers an untargeted, system-wide view of the transcriptome, enabling confirmation of on-target silencing and detection of potential unintended off-target effects [69] [70].
Quantitative Assessment of Silencing Efficiency
Large-scale analyses reveal critical trends in RNAi validation efficacy. A comprehensive 2016 study evaluating 429 siRNA experiments from 207 publications found that validation method choice significantly impacts measured silencing efficiency [69]. The data indicate that western blotting demonstrates the greatest apparent knockdown, followed by qPCR, with microarray analysis showing the most modest measured effects [69].
Table 1: RNAi Silencing Efficiency by Validation Method [69]
| Validation Method | Average Fold Change (mRNA Level) | Relative Performance |
|---|---|---|
| Western Blot | 0.43 ± 0.06 | Best |
| qPCR | 0.47 ± 0.10 | Intermediate |
| Microarray | 0.55 ± 0.06 | Least |
Cell line selection also significantly influences silencing outcomes. The same analysis found that among commonly used lines, SW480 colon cancer cells showed the best performance, while MCF7 breast cancer cells showed the lowest silencing efficiency [69].
Table 2: Cell Line Performance in RNAi Silencing [69]
| Cell Line | Type | Fold Change | Relative Efficiency |
|---|---|---|---|
| SW480 | Epithelial colon cancer | 0.30 ± 0.16 | Best |
| MDA-MB-231 | Breast cancer | 0.35 ± 0.20 | Intermediate |
| MCF7 | Breast cancer | 0.59 ± 0.06 | Least |
qRT-PCR Protocol for Silencing Confirmation
Sample Preparation and RNA Extraction
For cells in culture, begin by transfecting with target-specific siRNA (typically 1-30 nM) using appropriate transfection reagents [71] [72]. Include controls transfected with non-targeting siRNA. Harvest cells 24-72 hours post-transfection. Total RNA can be extracted using commercial kits (e.g., RNeasy Mini Kit) with on-column DNase digestion to remove genomic DNA contamination [71]. Alternatively, for rapid processing from limited cell numbers (as few as 3 cells), use Cells-to-Signal or similar kits that bypass RNA isolation through direct cell lysis [72].
cDNA Synthesis and qPCR Analysis
Synthesize cDNA from 1 μg total RNA using M-MLV reverse transcriptase with either random hexamers or oligo(dT)18 primers [71]. For SYBR Green-based qPCR, use Power SYBR Green Mastermix with primers designed to generate ~150 bp amplicons [71]. Carefully design primers to flank exon-exon junctions when using direct lysates to avoid genomic DNA amplification [72]. The following cycling conditions are recommended: 50°C for 10 minutes, 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute [71]. Analyze results using the ΔΔCt method, normalizing to housekeeping genes (e.g., β-actin, 18S rRNA) [71] [72].
Critical Considerations for qRT-PCR Validation
- Amplicon Position Matters: Primer placement significantly impacts knockdown detection. Amplicons located 5' to the siRNA cleavage site reliably detect silencing, while those 3' to the cleavage site may yield false negatives due to incomplete degradation of the 3' mRNA fragment [71].
- Specificity Controls: Run dissociation curves for SYBR Green assays to confirm single amplification products [72]. Include no-reverse-transcriptase controls to exclude genomic DNA contamination.
- Time Course Analysis: Measure mRNA levels at multiple time points (e.g., 24h and 72h post-transfection) as silencing dynamics vary [71].
Transcriptomic Approaches for Comprehensive Validation
Microarray Analysis Protocol
For genome-wide silencing assessment, extract high-quality total RNA with RIN (RNA Integrity Number) >8.5. Prepare labeled cRNA using standard protocols (e.g., Ambion RETROscript Kit) [1]. Hybridize to appropriate microarray platforms (e.g., Affymetrix GeneChip). Analyze data using robust multi-array average (RMA) normalization. Compare siRNA-treated samples to non-targeting siRNA controls to identify differentially expressed genes. Focus not only on the target gene but also on genome-wide patterns to detect potential off-target effects [69].
Data Interpretation and Quality Control
Successful silencing should show significant downregulation of the target gene (typical fold change <0.7) [69]. Examine off-target effects by identifying genes with sequence similarity to the siRNA seed region (nucleotides 2-8 of the guide strand) [69] [70]. Functional enrichment analysis of differentially expressed genes can reveal affected biological pathways. The high concordance between pre-designed siRNA libraries and commercial qPCR assays (e.g., Silencer siRNAs matched to TaqMan Gene Expression Assays) facilitates cross-platform validation [72].
Application in Fecundity and Egg Hatchability Research
In reproductive research, particularly insect pest control, RNAi targeting essential embryonic genes has demonstrated dramatic effects on fecundity and egg viability. For example, silencing the NlATG3 gene in brown planthopper (Nilaparvata lugens) resulted in complete mortality of 5th-instar nymphs and reduced egg hatchability from 95.7% to zero [2]. Similarly, targeting the Sl102 gene in Spodoptera littoralis eggs through dsRNA soaking significantly reduced embryonic survival and prevented larval hatching [1].
Table 3: RNAi Efficacy in Insect Fecundity Studies
| Target Gene | Species | Delivery Method | Effect on Fecundity | Effect on Hatchability |
|---|---|---|---|---|
| NlATG3 [2] | Nilaparvata lugens (brown planthopper) | Injection of dsRNA | Not specified | Reduced to 0% |
| Sl102 [1] | Spodoptera littoralis | Egg soaking in dsRNA | 80.4% reduction in eggs laid | Drastically reduced |
For embryonic studies, dsRNA delivery via egg soaking has proven effective. Protocol: collect synchronized eggs within 30 minutes of oviposition, soak in 50 μL PBS containing 250 ng/μL target-specific dsRNA for 120 minutes at 25°C [1]. Include control eggs soaked in dsRNA targeting non-functional genes (e.g., GFP). Monitor hatching rates and subsequent larval development [1].
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents for RNAi Validation Experiments
| Reagent | Function | Example Products |
|---|---|---|
| Pre-designed siRNAs | Ensure targeting efficacy and reproducibility | Silencer Pre-designed siRNAs (Ambion) [72] |
| RNA Extraction Kits | High-quality RNA isolation with DNA removal | RNeasy Mini Kit (Qiagen) [71] |
| Direct Lysis Kits | Rapid processing for high-throughput applications | Cells-to-Signal Kit (Ambion) [72] |
| Reverse Transcriptase | cDNA synthesis from RNA templates | M-MLV Reverse Transcriptase [71] |
| qPCR Master Mixes | Sensitive detection and quantification | Power SYBR Green Mastermix, TaqMan assays [71] [72] |
| Microarray Platforms | Genome-wide expression profiling | Affymetrix GeneChip [69] |
Robust molecular validation of RNAi-induced silencing is fundamental to research investigating fecundity and egg hatchability. qRT-PCR provides sensitive, quantitative confirmation of target gene knockdown, while transcriptomic approaches offer comprehensive assessment of specificity and off-target effects. The critical protocols and considerations outlined here provide researchers with a standardized framework for confirming RNAi efficacy, ensuring reliable results in both basic research and therapeutic development applications. Proper implementation of these validation strategies will accelerate progress in RNAi-based approaches for controlling reproduction across diverse biological systems.
The functional analysis of genes essential for reproduction, particularly those affecting fecundity and egg hatchability, relies heavily on precise gene silencing technologies. RNA interference (RNAi) and CRISPR-Cas9 represent two powerful but fundamentally distinct approaches for disrupting gene function. RNAi achieves targeted gene knockdown at the mRNA level through the introduction of double-stranded RNA (dsRNA), which triggers the degradation of complementary messenger RNA sequences [73]. In contrast, CRISPR-Cas9 facilitates permanent gene knockout at the DNA level by creating double-strand breaks that are repaired with insertions or deletions, disrupting the genetic code [73]. Understanding their temporal dynamics and functional outcomes is crucial for selecting the appropriate methodology in reproductive biology research, especially for investigating genes controlling insect fecundity and embryonic development.
Mechanism of Action and Temporal Dynamics
The fundamental difference in the level at which RNAi and CRISPR-Cas9 operate—transcriptional versus genetic—results in distinct temporal dynamics for phenotype manifestation, a critical consideration for experimental design in fecundity studies.
RNA Interference (RNAi): Transient Knockdown
- Process: Exogenously delivered dsRNA is processed by the Dicer enzyme into small interfering RNAs (siRNAs) of 21-25 nucleotides. These siRNAs are loaded into the RNA-induced silencing complex (RISC), which uses the antisense strand to identify and cleave complementary mRNA targets, preventing translation [73] [45] [74].
- Temporal Onset: Phenotypic changes following RNAi manifest relatively quickly, as the process directly reduces existing mRNA pools. Studies targeting reproductive regulators report observable impacts on fecundity and egg hatchability within the same generation after treatment [10].
- Reversibility: The effect is transient and reversible. As the dsRNA and siRNAs are degraded and new mRNA is transcribed, gene expression can recover, unless dsRNA is continuously supplied [73].
CRISPR-Cas9: Permanent Knockout
- Process: The CRISPR-Cas9 system consists of a Cas9 nuclease and a guide RNA (gRNA) that directs Cas9 to a specific DNA sequence. Upon binding, Cas9 creates a double-strand break in the DNA. The cell's repair mechanism, non-homologous end joining (NHEJ), often results in small insertions or deletions (indels) that disrupt the reading frame, leading to a permanent loss of gene function [73].
- Temporal Onset: Phenotypic manifestation is delayed compared to RNAi. The knockout is only realized after the mutated DNA is transcribed, which is contingent upon the turnover rate of the existing wild-type protein. In the context of reproduction, phenotypic consequences (e.g., reduced egg hatchability) may only be fully observable in the subsequent generation, after the germline has been edited [75].
- Permanence: The genetic change is heritable and permanent, allowing for the creation of stable mutant lines for ongoing study [73].
Table 1: Comparative Overview of RNAi and CRISPR-Cas9 Mechanisms
| Feature | RNAi (Knockdown) | CRISPR-Cas9 (Knockout) |
|---|---|---|
| Molecular Target | mRNA | DNA |
| Effect | Reduces gene expression | Disrupts the gene sequence |
| Temporal Onset | Relatively fast (hours to days) | Slower (days to generations) |
| Persistence | Transient and reversible | Heritable and permanent |
| Key Components | dsRNA/siRNA, Dicer, RISC, Argonaute | Cas9 Nuclease, Guide RNA (gRNA) |
Comparative Functional Outcomes in Fecundity and Egg Hatchability Research
Recent research targeting genes critical for insect reproduction highlights how the choice of technology influences experimental outcomes and interpretation.
Efficacy and Penetrance
- RNAi typically results in a partial reduction of the target protein. This knockdown can be sufficient to induce strong phenotypic effects. For instance, RNAi-mediated silencing of genes like tyrosine hydroxylase (TH) and dopa decarboxylase (DDC) in the small brown planthopper (Laodelphax striatellus) led to significantly shortened oviposition periods, reduced fecundity, and drastically inhibited egg hatchability [10]. Similarly, soaking eggs of Spodoptera littoralis in dsRNA targeting the Sl102 gene caused a drastic reduction in hatching rate and high mortality of hatched larvae [1].
- CRISPR-Cas9 aims for a complete and permanent loss of function. This is particularly valuable for studying essential genes where even residual protein activity can sustain some function. In the pea aphid (Acyrthosiphon pisum), CRISPR-Cas9 knockout of the Laccase2 (Lac2) gene, vital for eggshell sclerotization, resulted in completely unpigmented eggs, a significant loss of eggshell stiffness, and ultimately, embryonic lethality—a phenotype demonstrating the gene's absolute requirement for survival [75].
Specificity and Off-Target Effects
A critical consideration for attributing phenotypic outcomes to a specific gene target is the specificity of the technology.
- RNAi is historically more prone to off-target effects. These can occur when the siRNA unintentionally silences genes with partial sequence complementarity [73]. A comparative transcriptome analysis revealed that RNAi (using siRNAs) induced a larger number of deregulated mRNAs compared to both antibody-mediated loss-of-function and CRISPR-Cas9, suggesting a broader impact on the transcriptome that could confound phenotypic analysis [76].
- CRISPR-Cas9 offers high specificity, though off-target cleavage at near-identical DNA sequences can occur. This risk has been greatly mitigated through advanced gRNA design tools and the use of modified, high-fidelity Cas9 nucleases [73]. The same transcriptome study noted that CRISPR-Cas9 induced fewer off-target transcriptional changes than RNAi [76].
Table 2: Documented Phenotypic Outcomes in Insect Reproduction Studies
| Target Gene | Insect Species | Technology | Impact on Fecundity/Egg Hatchability | Source |
|---|---|---|---|---|
| Sl102 | Spodoptera littoralis | RNAi (dsRNA soaking) | Drastic reduction in egg hatching rate; high larval mortality. | [1] |
| TH & DDC | Laodelphax striatellus | RNAi (dsRNA injection/feeding) | Shortened oviposition, reduced fecundity, inhibited hatching & development. | [10] |
| Laccase2 | Acyrthosiphon pisum | CRISPR-Cas9 (Microinjection) | Complete loss of eggshell pigmentation, embryonic lethality, no hatching. | [75] |
Application Notes and Protocols for Reproductive Research
Selecting between RNAi and CRISPR-Cas9 depends on the experimental goal. RNAi is ideal for transient, reversible knockdown to study gene function in a specific life stage or to target essential genes whose complete knockout would be lethal. CRISPR-Cas9 is the tool of choice for creating stable, heritable knockouts to definitively establish gene function and for population-level studies.
Experimental Protocol: RNAi for Suppressing Egg Hatchability
This protocol details the dsRNA soaking method for targeting embryonic genes, adapted from successful studies in Lepidoptera [1].
Application: Functional analysis of genes critical for early embryonic development and egg hatchability. Principle: Permeation of dsRNA through the egg chorion to induce RNAi in the developing embryo. Key Considerations: Soaking duration, dsRNA concentration, and embryonic developmental stage are critical success factors [1].
Step-by-Step Workflow:
- dsRNA Preparation: Design and synthesize dsRNA targeting the gene of interest. A length of 200-500 bp is typically effective. A dsRNA targeting an unrelated gene (e.g., GFP) should be produced and used as a negative control.
- Egg Collection: Collect highly synchronized, newly laid eggs (within a 30-minute window) from the same egg mass to ensure uniform developmental stage.
- Soaking Treatment:
- Transfer approximately 120 eggs to a 1.5 mL microcentrifuge tube.
- Soak the eggs in 50 µL of dsRNA solution (e.g., 250 ng/µL in 1x PBS).
- Incubate at 25 ± 1°C for a determined duration (e.g., 120 minutes), with gentle agitation.
- Post-Treatment Incubation: After soaking, carefully remove the dsRNA solution, rinse the eggs if necessary, and transfer them to a fresh Petri dish with a suitable moist substrate.
- Phenotypic Assessment:
- Hatching Rate: Record the number of eggs that successfully hatch over time.
- Gene Silencing Efficiency: At desired time points, extract total RNA from eggs and use quantitative RT-PCR (qRT-PCR) to measure the transcript levels of the target gene.
- Morphological Analysis: Observe and document any developmental delays or morphological alterations in the embryos using structural and ultrastructural techniques [1].
Experimental Protocol: CRISPR-Cas9 for Germline Gene Knockout
This protocol outlines the use of CRISPR-Cas9 to create heritable mutations in genes affecting reproduction, based on refined methods for aphids [75].
Application: Generating stable mutant lines to study genes essential for fecundity, egg formation, and viability. Principle: Microinjection of Cas9 ribonucleoprotein (RNP) complexes into embryos or oviparous females to disrupt the target gene in the germline. Key Considerations: gRNA design and efficiency, timing of injection to target the germline, and overcoming species-specific biological challenges (e.g., symbiosis, high nuclease activity) are crucial [75].
Step-by-Step Workflow:
- gRNA Design and Synthesis: Identify a 20-nucleotide target sequence adjacent to a PAM (NGG for SpCas9) in an early exon of the target gene. Use design tools to predict efficiency and minimize off-targets. Synthesize the gRNA chemically or via in vitro transcription.
- RNP Complex Formation: Complex purified Cas9 protein with the synthesized gRNA to form the RNP complex. The RNP format reduces off-target effects and improves editing efficiency [73] [75].
- Microinjection:
- Option A (Early Embryo Injection): Inject the RNP complex into the cytoplasm of freshly laid (0-2 hour old) embryos.
- Option B (Direct Parental CRISPR/DIPA-CRISPR): Inject the RNP complex into the body cavity of adult oviparous females, targeting developing oocytes [75].
- Rearing and Screening (G0 Generation):
- Raise the injected individuals (G0) to adulthood. These are potential mosaics (crispants).
- For oviparous species, cross the G0 adults with wild-types and screen their offspring (G1) for the desired mutation.
- For species where DIPA-CRISPR is used, screen the eggs laid by the injected females (G0) directly [75].
- Genotyping and Line Establishment:
- Extract genomic DNA from individual G1 progeny or G0 eggs.
- Use PCR to amplify the target region and sequence the products to confirm the presence of indel mutations.
- Cross confirmed heterozygous mutants to establish stable homozygous knockout lines.
- Phenotypic Analysis:
- For fecundity/hatchability studies, assess reproductive parameters in the mutant line: number of eggs laid, egg morphology, pigmentation, stiffness, and hatching rate [75].
Diagram: Decision workflow for selecting RNAi or CRISPR-Cas9
The Scientist's Toolkit: Research Reagent Solutions
Successful implementation of RNAi and CRISPR-Cas9 protocols relies on key reagents and materials.
Table 3: Essential Research Reagents for Gene Silencing Studies
| Reagent/Material | Function | Example Application in Protocols |
|---|---|---|
| dsRNA (200-500 bp) | Triggers the RNAi pathway by serving as the precursor for siRNAs. | Soaking solution for insect eggs to silence embryonic genes [1]. |
| T7 or SP6 RNA Polymerase | Used for in vitro transcription to produce dsRNA. | Synthesis of dsRNA for RNAi experiments [1]. |
| Purified Cas9 Nuclease | The enzyme that creates double-strand breaks in DNA. | Component of the RNP complex for microinjection [75]. |
| Synthetic Guide RNA (gRNA) | Directs Cas9 to the specific genomic target site. | Component of the RNP complex; designed to target genes affecting reproduction [73] [75]. |
| Microinjection System | Precisely delivers reagents into embryos or adult insects. | Delivery of RNP complexes for CRISPR-Cas9 mutagenesis [75]. |
| Quantitative RT-PCR Kit | Quantifies mRNA levels to assess knockdown efficiency. | Validation of target gene silencing after RNAi treatment [1] [10]. |
Both RNAi and CRISPR-Cas9 are indispensable tools for functional genomics research aimed at reducing fecundity and egg hatchability. RNAi provides a rapid, flexible approach for transient knockdown, ideal for functional screening and stage-specific studies. CRISPR-Cas9 offers a definitive, permanent solution for establishing gene necessity and creating stable genetic models. The choice between them hinges on the experimental timeline, the desired persistence of the effect, and the specific biological question. A thorough understanding of their distinct temporal dynamics and functional outcomes ensures the correct application of these powerful technologies in advancing reproductive and developmental biology.
Within the context of a broader thesis focused on using RNA interference (RNAi) to reduce fecundity and egg hatchability in pest species, understanding the specificity and unintended consequences of gene silencing approaches is paramount. While RNAi has emerged as a powerful tool for functional genomics and pest control, its application can be confounded by off-target effects (OTEs), where non-target genes are inadvertently silenced. This application note provides a comparative analysis of RNAi and an alternative approach—antibody-mediated loss-of-function—focusing on their transcriptome-wide off-target signatures. We present standardized protocols for assessing these effects, enabling researchers to design more reliable and interpretable experiments aimed at compromising reproductive success in target organisms.
Comparative Analysis of Techniques and Off-Target Effects
Mechanism of Action and Key Characteristics
Table 1: Comparison of RNAi and Antibody-Mediated Knockdown Mechanisms
| Feature | RNA Interference (RNAi) | Antibody-Mediated Loss-of-Function |
|---|---|---|
| Fundamental Mechanism | Post-transcriptional gene silencing via mRNA degradation or translational inhibition [77] [78] | Direct intracellular antibody-target protein interaction; phenotypic change without altering mRNA/protein levels [76] |
| Key Effector Molecules | Double-stranded RNA (dsRNA), small interfering RNA (siRNA), short hairpin RNA (shRNA) [78] [79] | Monoclonal or recombinant antibodies delivered intracellularly [76] |
| Effect on Target | Knock-down (reduction) of target mRNA and protein [78] | Functional blockade of the target protein's activity [76] |
| Temporal Onset of Phenotype | Delayed; requires turnover of existing mRNA/protein [76] | Relatively rapid; direct inhibition of protein function [76] |
| Primary Application | Large-scale functional genomics, therapeutic gene silencing, pest population control [77] [79] [8] | Target validation, functional complementation of genetic studies, modulating protein activity [76] |
Quantitative Profile of Transcriptome-Wide Off-Target Effects
A direct comparative study analyzing changes in cell adhesion by targeting Talin1 (TLN1) and Kindlin-2 provides critical, quantitative insights into the off-target profiles of these methodologies [76].
Table 2: Transcriptome-Wide Off-Target Effect Profile
| Method | Number of Deregulated mRNAs (Relative to Control) | Overlap of Deregulated Transcripts with Negative Control |
|---|---|---|
| RNAi (siRNA) | Highest number | ~10% [76] |
| CRISPR-Cas9 Knock-out | Fewer than RNAi | ~70% [76] |
| Antibody-Mediated Loss-of-Function | Fewer than RNAi | ~30% [76] |
The data indicates that antibody-mediated knockdown induces a transcriptomic off-target profile distinct from both RNAi and CRISPR-Cas9, with a significantly lower overlap with its negative control compared to CRISPR-Cas9, suggesting a different mechanism of action. RNAi demonstrates the most divergent off-target signature, with the lowest overlap with its control, highlighting its potential for unintended transcriptomic changes.
Experimental Protocols
Protocol 1: Standardized RNAi Knockdown for Fecundity Research
This protocol is adapted from successful RNAi experiments that disrupted egg development in the pest insect Locusta migratoria by targeting essential genes [8].
Key Reagent Solutions
- dsRNA Synthesis Kit: For in vitro transcription of target gene dsRNA (e.g., targeting a gene vital for embryogenesis like LmGFAT in locusts) [8].
- Nuclease-Free Water: For resuspending and diluting dsRNA [8].
- Chitosan Nanopolymer: A delivery enhancer that complexes with dsRNA to protect it from degradation and improve cellular uptake [8].
- PBS Buffer: For control injections and sample preparation [8].
Step-by-Step Procedure
dsRNA Preparation and Purification
- Design: Select a 400-500 bp target sequence from the gene of interest (e.g., LmGFAT). Perform a specificity check via BLAST to minimize off-target potential [8].
- Synthesize: Produce dsRNA in vitro using a commercial kit. A critical quality control step is the complete removal of DNA template through DNase I treatment and phenol/chloroform extraction, followed by ethanol precipitation. Incomplete purification drastically reduces knockdown efficiency [80].
- Quantify: Measure dsRNA concentration using a spectrophotometer and confirm integrity via agarose gel electrophoresis [8].
Delivery of dsRNA
- Complexation (Optional but Recommended): For enhanced efficacy, complex dsRNA with chitosan. Mix dsRNA with chitosan solution at an optimal mass ratio (e.g., 1:5 to 1:20 dsRNA:chitosan) and incubate at room temperature for 30 minutes to form stable nanoparticles [8].
- Administration: For insect egg/embryo knockdown, use microinjection. Inject 500 nL of purified dsRNA (e.g., at 1 µg/µL) or chitosan-complexed dsRNA directly into the embryo. A control group should be injected with dsRNA targeting a non-functional gene (e.g., GFP) [8].
Phenotypic and Molecular Assessment
- Monitor Phenotype: Incubate injected eggs and record egg mortality rates and hatchability over time. For locust LmGFAT knockdown, a >95% egg developmental arrest is observed [8].
- Validate Knockdown Efficiency: After phenotypic observation, extract total RNA from a sample of treated and control embryos/individuals. Perform RT-qPCR to quantify the reduction in target mRNA levels [8] [80].
Protocol 2: Antibody-Mediated Loss-of-Function and Off-Target Assessment
This protocol is based on a comparative study that used antibody transfection to modulate cell adhesion proteins [76].
Key Reagent Solutions
- Target-Specific Monoclonal Antibodies: Antibodies with high affinity and specificity for the target protein [76].
- Transfection Reagent: Suitable for intracellular antibody delivery into cultured cells [76].
- Cell Culture Medium: Appropriate for maintaining the target cell line [76].
- RNA Lysis Buffer: For subsequent RNA extraction for transcriptome analysis [76].
Step-by-Step Procedure
Antibody Preparation and Transfection
- Antibody Selection: Procure or generate highly specific monoclonal antibodies against the target protein.
- Complex Formation: Complex the antibodies with the transfection reagent according to the manufacturer's protocol to form antibody-loaded particles [76].
- Cell Transfection: Apply the antibody-transfection complexes to cultured cells. Include control groups transfected with non-targeting IgG or transfection reagent alone [76].
Phenotypic Validation
- Functional Assay: Perform relevant phenotypic assays (e.g., cell adhesion assay, observation of egg development in in vitro models) at 24-48 hours post-transfection. Antibody-mediated knockdown typically results in a rapid onset of phenotypic changes [76].
Transcriptome-Wide Off-Target Analysis
- RNA Extraction: At the experimental endpoint, extract total RNA with high purity from treated and control cells using a commercial kit [76] [77].
- Transcriptome Sequencing: Prepare RNA-seq libraries and perform high-throughput sequencing on a platform such as Illumina. Sequence to a sufficient depth (e.g., 30 million reads per sample) for robust transcript quantification [76] [77].
- Bioinformatic Analysis:
- Map sequencing reads to the reference genome/transcriptome.
- Quantify gene expression levels.
- Identify Differentially Expressed Genes (DEGs) by comparing treated samples to controls (e.g., using a threshold of |log2 fold-change| > 1 and adjusted p-value < 0.05).
- Perform Gene Ontology (GO) enrichment analysis on the DEG lists to identify affected biological processes [76] [77].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Knockdown and OTE Analysis
| Reagent | Function | Application Notes |
|---|---|---|
| dsRNA Synthesis Kit | In vitro production of dsRNA triggers for RNAi. | Ensure high-yield synthesis. Critical step is thorough DNA template removal [80]. |
| Chitosan | Natural polysaccharide nanoparticle that complexes with and protects dsRNA. | Enhances RNAi efficiency, especially in recalcitrant species, by improving cellular uptake [8]. |
| Microinjection System | Precision delivery of dsRNA or antibodies into small organisms or embryos. | Essential for targeting specific tissues or early developmental stages [8]. |
| Monoclonal Antibodies | Highly specific binders for antibody-mediated functional blockade. | Must be validated for intracellular activity post-transfection [76]. |
| Intracellular Transfection Reagent | Enables delivery of antibodies (and other macromolecules) into the cell cytoplasm. | Compatibility with antibodies and cell viability are key selection criteria [76]. |
| RNA Extraction Kit | Isolation of high-purity, intact total RNA. | Purity (A260/280 > 2.0) is critical for downstream transcriptomic applications [76] [77]. |
| RNA-seq Library Prep Kit | Preparation of sequencing libraries from total RNA. | Select kits that preserve information on strand orientation and low-input samples [76]. |
Choosing between RNAi and antibody-mediated knockdown requires a strategic balance between experimental goals, model system, and the imperative to minimize off-target effects. For large-scale functional genetic screens or applications like pest control targeting fecundity, RNAi remains a potent tool, though its significant and distinct off-target signature necessitates careful control design and validation. In contrast, antibody-mediated loss-of-function presents a valuable alternative for target validation and precise functional studies, offering a different mechanism of action with a potentially more favorable transcriptome-wide off-target profile. By employing the standardized protocols and analytical frameworks outlined here, researchers can more critically assess the specificity of their interventions, leading to more robust and interpretable conclusions in their research on fecundity and developmental biology.
Conclusion
The strategic application of RNAi to reduce fecundity and egg hatchability presents a powerful and targeted approach for pest and vector control. By silencing critical genes involved in embryogenesis, neuroendocrine function, and structural development, RNAi induces significant reproductive failure and early mortality. While challenges in delivery and efficiency persist, advancements in formulation, dsRNA design, and high-throughput screening are paving the way for more robust applications. Future research should focus on improving in vivo stability, exploring combination therapies with other biocontrol agents, and expanding the scope of target organisms. This methodology not only offers a sustainable alternative to chemical pesticides but also holds promise for novel therapeutic strategies, positioning RNAi as a cornerstone of next-generation precision biological control.
References
- 1. RNAi-mediated suppression of embryos as a promising strategy to control Spodoptera littoralis | Journal of Pest Science [https://link.springer.com/article/10.1007/s10340-025-01932-z]
- 2. RNAi-mediated silencing of the autophagy-related gene ... [https://pubmed.ncbi.nlm.nih.gov/34092014/]
- 3. Knockdown of RNA interference pathway genes impacts ... [https://www.nature.com/articles/s41598-018-26129-6]
- 4. Dopamine synthesis and dopamine receptor expression ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5958746/]
- 5. High-Throughput Selection of Effective RNAi Probes for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC403718/]
- 6. Are There Connections between the Neuroendocrine and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12453576/]
- 7. Validation of RNAi Silencing Efficiency Using Gene Array ... [https://www.sciencedirect.com/science/article/pii/S2162253117300859]
- 8. Disruption of egg and nymph development via RNAi ... [https://www.sciencedirect.com/science/article/abs/pii/S004835752500272X]
- 9. The CYP303A1 is essential in embryonic development of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12540425/]
- 10. RNA interference targeting tyrosine hydroxylase and dopa ... [https://www.sciencedirect.com/science/article/abs/pii/S0048357525004766]
- 11. Protocol for RNA interference in gene expression in adult ... [https://www.sciencedirect.com/science/article/pii/S2666166725002710]
- 12. RNA interference knockdown of the nuclear receptor HR3 ... [https://pubmed.ncbi.nlm.nih.gov/40397197/]
- 13. FlyRNAi.org 2025 update—expanded resources for new ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11701652/]
- 14. Double-stranded RNA delivery through soaking stimulates ... [https://www.sciencedirect.com/science/article/abs/pii/S0044848625005678]
- 15. Protocol combining RNA interference and regeneration ... [https://pubmed.ncbi.nlm.nih.gov/41273725/]
- 16. RNAi-mediated knockdown of juvenile hormone acid ... [https://pubmed.ncbi.nlm.nih.gov/40350233/]
- 17. Successful oral RNA interference efficiency in the silkworm ... [https://www.sciencedirect.com/science/article/abs/pii/S0022191025000034]
- 18. Investigating Engineered Ribonucleoprotein Particles to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5408074/]
- 19. Aptamer-conjugated gold nanoparticles enable ... [https://www.nature.com/articles/s41467-024-55223-9]
- 20. RNA Therapeutics: Delivery Problems and Solutions—A Review [https://pmc.ncbi.nlm.nih.gov/articles/PMC12567017/]
- 21. Optimizing dsRNA sequences for RNAi in pest control and ... [https://bmcbiol.biomedcentral.com/articles/10.1186/s12915-025-02219-6]
- 22. RNAi in Pest Control: Critical Factors Affecting dsRNA ... [https://www.mdpi.com/2075-4450/16/7/737]
- 23. RNAi revolution in agriculture: unlocking mechanisms ... [https://pubmed.ncbi.nlm.nih.gov/40629260/]
- 24. Evaluation of RNA isolation techniques for enhanced ... [https://pubmed.ncbi.nlm.nih.gov/40970954/]
- 25. New Technique Silences Plant Genes to Boost Crop Yields [https://www.seedworld.com/europe/2025/08/18/new-technique-silences-plant-genes-to-boost-crop-yields/]
- 26. RNA Interference Drug Delivery Market Size 2025 to 2034 [https://www.precedenceresearch.com/rna-interference-drug-delivery-market]
- 27. Hurdles to healing: Overcoming cellular barriers for viral ... [https://www.sciencedirect.com/science/article/pii/S0378517325003060]
- 28. Parental RNA interference (pRNAi) in insects [https://pubmed.ncbi.nlm.nih.gov/40531353/]
- 29. Viral and non-viral vectors in gene therapy: current state ... [https://www.sciencedirect.com/science/article/pii/S2352396425002786]
- 30. Unleashing the potential of mRNA: Overcoming delivery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11883111/]
- 31. Past, Present, and Future of Viral Vector Vaccine Platforms [https://pmc.ncbi.nlm.nih.gov/articles/PMC12115715/]
- 32. Leucine aminopeptidase1 controls egg deposition and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10762072/]
- 33. Viral vectors: design and delivery for small RNA [https://pubmed.ncbi.nlm.nih.gov/39950625/?utm_source=FeedFetcher&utm_medium=rss&utm_campaign=None&utm_content=1h5ksaKdwjSVxnNNwl5mj_zu4UUlqukdhnrNuWLIVwmhtqDgLq&fc=None&ff=20251104060059&v=2.18.0.post22+67771e2]
- 34. Protocol Knock down of target genes by RNA interference ... [https://www.sciencedirect.com/science/article/pii/S2666166722000995]
- 35. RNAi mediated knockdown of RpL11, RpS2, and tra-2 led ... [https://pubmed.ncbi.nlm.nih.gov/31375950/]
- 36. High Throughput Screening Market Forecast, 2025-2032 [https://www.coherentmarketinsights.com/industry-reports/high-throughput-screening-market]
- 37. High-throughput Screening of HiBiT-tagged Proteins [https://ginkgo.bio/resources/application-notes/high-throughput-screening-hibit-proteins]
- 38. Protocol for high-throughput screening of SIRT7 inhibitors ... [https://www.sciencedirect.com/science/article/pii/S2666166725001261]
- 39. The role of polymers in enabling RNAi-based technology ... [https://www.nature.com/articles/s41467-024-53468-y]
- 40. Effects of Double-Stranded RNA Degrading Nucleases on ... [https://www.mdpi.com/2075-4450/16/2/229]
- 41. Increased and synergistic RNAi delivery using MOF ... [https://www.nature.com/articles/s41467-025-61604-5]
- 42. A novel self-assembled nanocarrier-mediated dsRNA ... [https://www.sciencedirect.com/science/article/pii/S2590006425006696]
- 43. Double‐Stranded DNA Reduces dsRNA Degradation in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12447125/]
- 44. Overcoming soil matrix interference for reliable ... [https://www.sciencedirect.com/science/article/pii/S2352186425003694]
- 45. RNAi in Pest Control: Critical Factors Affecting dsRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12295715/]
- 46. Cryo-EM structure reveals how SID-1 recognizes dsRNA ... [https://www.nature.com/articles/s41594-024-01277-8]
- 47. Structural insight into the human SID1 transmembrane ... [https://www.nature.com/articles/s41467-023-39335-2]
- 48. Molecular mechanisms of dsRNA uptake and intracellular ... [https://www.sciencedirect.com/science/article/abs/pii/S0965174825001766]
- 49. Structural basis for double-stranded RNA recognition by SID1 [https://pmc.ncbi.nlm.nih.gov/articles/PMC11194109/]
- 50. RNA Interference–Based Therapy and Its Delivery Systems [https://pmc.ncbi.nlm.nih.gov/articles/PMC5898634/]
- 51. RNAI-based strategies and nanomaterial-mediated ... [https://www.sciencedirect.com/science/article/pii/S2090123225008392]
- 52. Optimizing dsRNA sequences for RNAi in pest control and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12039203/]
- 53. RNA Interference: A Potent Tool for Gene‐Specific Therapeutics [https://pmc.ncbi.nlm.nih.gov/articles/PMC7175948/]
- 54. siRMSD: A Structural Parameter to Reduce Sequence ... [https://pubmed.ncbi.nlm.nih.gov/40877521/]
- 55. Engineered mRNAs With Stable Structures Minimize ... [https://www.sciencedirect.com/science/article/abs/pii/S0022283624004443]
- 56. a web-based tool of comprehensive dsRNA design for pest ... [https://www.sciencedirect.com/science/article/abs/pii/S0167779925000022]
- 57. An RNA Interference Tool to Silence Genes in Sarcoptes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8777771/]
- 58. Identification of highly effective target genes for RNAi ... [https://www.nature.com/articles/s41598-018-23216-6]
- 59. Nanocarrier-mediated RNAi of CYP9A306 and CYB5R ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1573634/full]
- 60. RNAi-Based Therapeutics and Novel RNA Bioengineering ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9827509/]
- 61. Systematic analysis of siRNA and mRNA features impacting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12205987/]
- 62. Roles of Vitellogenin and Its Receptor Genes in Female Reproduction of the Cigarette Beetle, Lasioderma serricorne [https://www.mdpi.com/2075-4450/16/2/175]
- 63. RNA interference screening demystified - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2652024/]
- 64. Nanoclay-Mediated RNA Interference Targeting Insecticide Receptors Enhances Whitefly Mortality and Reduces Fecundity | Molecular Biotechnology [https://link.springer.com/article/10.1007/s12033-025-01529-y]
- 65. Silencing of the BtTPS genes by transgenic plant-mediated RNAi to control Bemisia tabaci MED - PubMed [https://pubmed.ncbi.nlm.nih.gov/34796637/]
- 66. In Vivo RNAi Protocols [https://www.thermofisher.com/cl/en/home/life-science/rnai/introduction-to-in-vivo-rnai/in-vivo-rnai-protocols.html]
- 67. Comparison of siRNA-induced off-target RNA and protein effects [https://pmc.ncbi.nlm.nih.gov/articles/PMC1800510/]
- 68. Gene Specific Silencing by RNAi [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/rnai-sirna/tech-notes/gene-specific-silencing-by-rnai.html]
- 69. Validation of RNAi Silencing Efficiency Using Gene Array ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5056990/]
- 70. Classification of and detection techniques for RNAi ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1535384/full]
- 71. Detection of siRNA induced mRNA silencing by RT-qPCR [https://pmc.ncbi.nlm.nih.gov/articles/PMC2850347/]
- 72. Fast Accurate Assessment of siRNA-induced Gene Silencing [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/rnai-sirna/tech-notes/fast-accurate-assessment-of-sirna-induced-gene-silencing.html]
- 73. RNAi vs. CRISPR: Guide to Selecting the Best Gene ... [https://www.synthego.com/blog/rnai-vs-crispr-guide/]
- 74. RNAi turns 25:contributions and challenges in insect science [https://www.frontiersin.org/journals/insect-science/articles/10.3389/finsc.2023.1209478/full]
- 75. Refined CRISPR/Cas9 genome editing in the pea aphid ... [https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1011557]
- 76. Comparative analysis of antibody-mediated loss ... [https://www.sciencedirect.com/science/article/pii/S2472555225000760]
- 77. Assessment of Potential Toxic Effects of RNAi-Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12467772/]
- 78. RNA interference: learning gene knock-down from cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC534783/]
- 79. Advances in RNA-based therapeutics: current ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1675209/full]
- 80. RNAi-directed knockdown in the cnidarian fish blood ... [https://www.nature.com/articles/s41598-024-54171-0]