Sperm Epigenetics Decoded: A Comprehensive Guide to the Infinium Methylation BeadChip
This article provides a comprehensive resource for researchers and drug development professionals utilizing the Illumina Infinium Methylation BeadChip for sperm epigenetics studies.
Sperm Epigenetics Decoded: A Comprehensive Guide to the Infinium Methylation BeadChip
Abstract
This article provides a comprehensive resource for researchers and drug development professionals utilizing the Illumina Infinium Methylation BeadChip for sperm epigenetics studies. We cover the foundational principles of sperm DNA methylation and its link to offspring health, detail methodological best practices from sample collection to data analysis, and offer troubleshooting guidance for common technical challenges. Furthermore, we critically evaluate the platform's performance, including comparisons with sequencing technologies and assessments of the latest EPICv2 array, to empower robust and reproducible research into the paternal germline's role in development and disease.
The Paternal Germline Blueprint: Linking Sperm DNA Methylation to Offspring Health
Sperm epigenetics represents a critical field of study examining the molecular processes that regulate gene expression without altering the underlying DNA sequence in male gametes. The sperm epigenome is characterized by a unique DNA methylation landscape that is fundamentally distinct from somatic cells, establishing a specific chromatin architecture essential for proper embryo development [1]. Research has demonstrated that this epigenetic state is not static but dynamically influenced by various factors including paternal aging, environmental toxin exposure, and lifestyle factors such as obesity, with significant implications for sperm fertility and the health trajectory of offspring [2] [3].
The growing interest in this field stems from the epigenetic mechanism's role as a potential mediator between environmental exposures and phenotypic outcomes in subsequent generations. Notably, children of aged fathers have been documented to be at a higher risk for various neurodevelopmental disorders and mental health conditions, with alterations in sperm DNA methylation patterns proposed as a contributing biological mechanism [1]. Understanding these dynamics provides valuable insights into transgenerational inheritance and offers potential diagnostic and therapeutic avenues for male factor infertility.
The Infinium Methylation BeadChip Platform
The Infinium Methylation BeadChip, manufactured by Illumina, is a microarray-based technology designed for robust, genome-wide DNA methylation analysis. This platform has become one of the most widely used technologies in epigenome-wide association studies (EWAS) due to its cost-effectiveness, high accuracy, and user-friendly data analysis pipelines compared to sequencing-based methods [4] [5]. The technology utilizes two different probe designs (Infinium I and Infinium II) to quantify methylation status at cytosine residues within CpG dinucleotides following bisulfite conversion of DNA [4].
The platform has evolved through several generations, each expanding genomic coverage. The most recent iteration, the Infinium MethylationEPIC v2 BeadChip (EPICv2), features 937,690 probes and offers significant improvements over its predecessors, including enhanced coverage of enhancer regions, applicability to diverse ancestry groups, and support for low-input DNA down to one nanogram [4]. The array provides balanced coverage across key genomic regions including CpG islands, translation start sites, enhancers, and imprinted loci, enabling comprehensive epigenetic profiling [6].
Table 1: Evolution of Infinium Human Methylation BeadChips
| Array Version | Number of Probes | Key Features | Low Input DNA Support |
|---|---|---|---|
| HM27 | ~27,000 | Focus on promoter regions | Not specified |
| HM450 | ~486,427 | Expansion to gene body methylation | Not specified |
| EPIC v1 | ~866,552 | Enhanced coverage of enhancer regions | Not specified |
| EPIC v2 | ~937,690 | Improved probe mapping, diverse ancestry applicability, somatic mutation targets | 1 ng |
Technical performance metrics demonstrate the platform's reliability, with the EPICv2 achieving >98% reproducibility for technical replicates and high correlation with whole-genome bisulfite sequencing data [6] [4]. The technology's quantitative performance, combined with its relatively low DNA input requirements and high-throughput capacity, makes it particularly suitable for large cohort studies in both clinical and research settings.
Key Applications in Sperm Epigenetics Research
Paternal Aging and Sperm Quality
The investigation of age-related epigenetic alterations in sperm represents a major application of the Infinium BeadChip platform. Advanced paternal age has been associated with increased risk for neurodevelopmental disorders in offspring, and DNA methylation changes in sperm are hypothesized as a potential mechanism [1]. Using a customized methylC-capture sequencing approach validated against array data, researchers identified more than 150,000 age-related CpG sites in sperm, with a predominance of hypermethylation (62%) compared to hypomethylation (38%) in aged men [1].
These age-associated epigenetic changes are not randomly distributed across the genome. Hypermethylated sites in aged sperm are frequently located in distal gene regions, while hypomethylated sites tend to occur near transcription start sites [1]. Particularly dense clusters of age-related changes have been identified on chromosomes 4 and 16, with the chromosome 4 cluster overlapping the PGC1α locus (involved in metabolic aging) and the chromosome 16 cluster overlapping the RBFOX1 locus (implicated in neurodevelopmental disease) [1]. Gene ontology analyses reveal that genes most affected by age-associated methylation changes are enriched for biological processes related to development, neuron projection, differentiation, and behavior [1].
Table 2: Sperm Age Prediction Models Using Methylation Arrays
| Study | Technology | Key Markers/Regions | Prediction Accuracy (MAE) |
|---|---|---|---|
| Jenkins et al. [1] | 450K array | 139 hypomethylated, 8 hypermethylated regions | Not specified |
| Lee et al. [7] [8] | 450K array | TTC7B, FOLH1B, LOC401324 | 5.4 years (3-marker model) |
| Pisarek et al. [8] | EPIC array | SH2B2, EXOC3, IFITM2, GALR2, FOLH1B | 5.1 years (6-marker model) |
| Current Study [1] | MCC-seq | >150,000 CpGs | Improved accuracy over 450K |
Figure 1: Pathway of Paternal Aging Effects on Sperm Epigenetics and Offspring Health
Forensic Applications
DNA methylation analysis in semen has significant applications in forensic science, particularly for age prediction from evidence collected at crime scenes. Semen samples are frequently encountered in sexual assault cases, and accurate age estimation can provide valuable investigative leads when conventional DNA profiling fails to identify a suspect [7] [8]. The Infinium BeadChip platform has been instrumental in identifying semen-specific age-related methylation markers.
Research comparing methylation patterns between European and Korean populations has revealed significant population-specific differences in age-related methylation markers, necessitating the development of population-tailored prediction models [7]. This highlights the importance of considering genetic ancestry in forensic epigenetic applications. Recent studies utilizing the Infinium MethylationEPIC BeadChip have identified novel age-associated markers that improve prediction accuracy compared to earlier models based on the 450K array [7] [8].
Environmental and Lifestyle Factors
The Infinium BeadChip platform has also facilitated investigations into how lifestyle factors such as obesity interact with paternal aging to influence the sperm epigenome. Although one study found no statistically significant epigenetic age acceleration associated with high BMI, researchers observed a consistent trend where individuals with high BMI were predicted to be epigenetically older than their chronological age across all age categories [3]. When BMI was included as a feature in age prediction models, a modest non-significant improvement in predictive accuracy was observed (r² = 0.8814, MAE = 3.2913 with BMI vs. r² = 0.8739, MAE = 3.3567 without BMI) [3].
Additionally, studies have examined the impact of environmental toxin exposure on sperm DNA methylation, with implications for sperm DNA quality and fertility [2]. These investigations leverage the comprehensive epigenome coverage provided by the Infinium platform to identify potential mechanistic links between environmental exposures and reproductive health outcomes.
Experimental Protocols and Methodologies
Sample Preparation and Quality Control
Proper sample preparation is crucial for generating reliable sperm methylation data. A comprehensive approach to addressing somatic DNA contamination in sperm epigenetic studies includes both pre-analytical and analytical steps [2]:
- Microscopic Examination: Initial visual inspection to assess sample purity.
- Somatic Cell Lysis Buffer (SCLB) Treatment: Chemical treatment to selectively lyse somatic cells while preserving sperm integrity.
- Bisulfite Pyrosequencing of Imprinted Loci: Analytical verification using loci with known methylation patterns in sperm versus somatic cells. The DLK1 locus, which is highly methylated in somatic cells but unmethylated in sperm, serves as a reliable discriminator [3].
- Infinium Array Data Analysis: Computational assessment using 9,564 CpG sites identified as highly methylated in blood compared to sperm, serving as contamination biomarkers [2].
Studies recommend applying a 15% cutoff during data analysis to completely eliminate the influence of somatic DNA contamination in sperm epigenetic studies [2]. This comprehensive quality control protocol ensures that observed methylation patterns truly reflect the sperm epigenome rather than contamination from somatic cells.
Sperm Epigenetic Age Prediction Workflow
The following workflow outlines a standardized protocol for developing sperm-specific age prediction models using the Infinium BeadChip platform:
Figure 2: Workflow for Sperm Epigenetic Age Prediction Using Infinium BeadChip
Cohort Selection: Recruit donors across a broad age range (e.g., 18-70 years) with appropriate ethical approvals [7]. Sample sizes in recent studies have ranged from 94 to 161 individuals [7] [1].
DNA Extraction and Bisulfite Conversion: Extract genomic DNA from semen samples using standardized protocols. Convert DNA using bisulfite treatment (e.g., EZ DNA Methylation Kit) to convert unmethylated cytosines to uracils while leaving methylated cytosines unchanged [8].
Array Processing: Process 250 ng of bisulfite-converted DNA on the Infinium MethylationEPIC BeadChip according to manufacturer's protocols, followed by scanning on an iScan System [6] [4].
Data Preprocessing: Process raw .idat files using specialized bioinformatics tools such as SeSAMe, minfi, or ChAMP to perform background subtraction, control normalization, and quality assessment [5] [9]. Remove probes containing SNPs at the CpG interrogation or single-nucleotide extension sites to minimize genetic confounding [5].
Marker Selection and Model Building: Identify age-associated CpG sites using correlation analysis (e.g., Pearson's r with p < 0.00001) and false discovery rate correction (FDR ≤ 0.05) [8]. Develop prediction models using multivariable linear regression on power-transformed DNA methylation data, supported by Bayesian Information Criterion for marker selection [8].
Model Validation: Validate prediction models in independent sample sets to assess performance metrics including mean absolute error (MAE) and correlation between predicted and chronological age [7] [8].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents and Materials for Sperm Epigenetics Studies
| Item | Function | Specifications |
|---|---|---|
| Infinium MethylationEPIC BeadChip Kit | Genome-wide DNA methylation profiling | 8 samples per array, >850,000 CpG sites, 250 ng DNA input |
| Somatic Cell Lysis Buffer | Selective removal of somatic cells from semen samples | Preserves sperm integrity while lysing contaminating cells |
| Bisulfite Conversion Kit | Converts unmethylated cytosine to uracil for methylation detection | Enables discrimination of methylated/unmethylated sites |
| iScan System | Array scanning and imaging | Fluorescent detection of hybridized arrays |
| DLK1 Imprinted Locus Assay | Sperm purity assessment via pyrosequencing | Validates absence of somatic contamination |
| SeSAMe Software Package | Bioinformatics analysis of array data | Quality control, normalization, differential methylation |
| GenomeStudio Methylation Module | Initial data quality assessment | Control probe visualization, basic QC analysis |
The Infinium Methylation BeadChip platform represents a powerful tool for advancing sperm epigenetics research, offering comprehensive coverage of the dynamic sperm methylome with robust technical performance. Applications in studying paternal aging, forensic age prediction, and environmental influences demonstrate the platform's versatility across basic, clinical, and forensic research domains. The continued refinement of experimental protocols—particularly for addressing somatic cell contamination and accounting for population-specific methylation patterns—will further enhance the reliability and applicability of findings in this rapidly evolving field. As research progresses, the integration of methylation array data with other multi-omics approaches promises to provide unprecedented insights into the role of sperm epigenetics in inheritance and offspring health.
This document provides Application Notes and Protocols for investigating key biological processes—spermatogenesis, genomic imprinting, and environmental response—within the context of sperm epigenetics research using the Infinium MethylationEPIC v2 BeadChip. This platform enables cost-effective, quantitative, and user-friendly genome-wide profiling of DNA cytosine modifications, which are critical for understanding male fertility, transgenerational inheritance, and the epigenetic impacts of environmental stressors [4]. These notes are designed to guide researchers and drug development professionals in applying this technology to explore the epigenetic regulation of sperm function.
Biological Foundations and Epigenetic Significance
Spermatogenesis: The Production of Male Gametes
Spermatogenesis is the complex, multi-stage process through which haploid spermatozoa develop from germ cells in the seminiferous tubules of the testes. It is crucial for sexual reproduction, ensuring the production of genetically unique, mobile gametes capable of fertilizing an oocyte [10] [11].
Key Stages and Cellular Transformations: The process begins at puberty and continues uninterrupted throughout life, taking approximately 72-74 days in humans [11]. It can be divided into three key phases:
- Spermatocytogenesis: Spermatogonial stem cells undergo mitotic divisions to both self-renew and produce diploid primary spermatocytes [10] [11].
- Meiosis: Primary spermatocytes undergo two reductive divisions (Meiosis I and II) to generate haploid spermatids. This introduces genetic variation through chromosomal crossover and random assortment [10] [11].
- Spermiogenesis: A final maturation phase where round spermatids undergo extensive morphological transformation into mature, motile spermatozoa. This involves nuclear condensation via histone-to-protamine exchange, acrosome formation, and flagellum development [10] [12].
Table: Stages of Spermatogenesis and Key Characteristics
| Cell Type | Ploidy | DNA Copy Number | Primary Process | Key Epigenetic Event |
|---|---|---|---|---|
| Spermatogonium | Diploid (2N) / 46 | 2C / 46 | Mitosis | -- |
| Primary Spermatocyte | Diploid (2N) / 46 | 4C / 2x46 | Meiosis I | Homologous recombination |
| Secondary Spermatocyte | Haploid (N) / 23 | 2C / 2x23 | Meiosis II | -- |
| Spermatid | Haploid (N) / 23 | C / 23 | Spermiogenesis | Histone-to-protamine exchange, transcriptional shutdown |
| Spermatozoon | Haploid (N) / 23 | C / 23 | Spermiation | Fully packaged, transcriptionally silent genome |
The developing germ cells are supported by Sertoli cells, which provide structural support, nutrition, and form the blood-testis barrier, creating a protected microenvironment for spermatogenesis [10] [11]. Leydig cells, located in the inter-tubular space, produce testosterone, which is essential for initiating and maintaining the process [10].
Epigenetic Reprogramming in Sperm: Sperm is epigenetically programmed to regulate gene expression in the embryo [13]. During spermiogenesis, the nucleus undergoes dramatic compaction where most histones are replaced by protamines. However, approximately 1-10% of histones are retained, particularly at promoters of developmentally important genes [13]. These retained histones carry post-translational modifications (e.g., H3K4me2/3, H3K27me3) that are hypothesized to deliver epigenetic instructions to the zygote, potentially influencing embryonic transcription and development [13]. This epigenetic signature makes sperm a critical vector for paternal environmental exposures and a subject of intense study in transgenerational inheritance.
Genomic Imprinting: Parental-Origin Specific Gene Expression
Genomic imprinting is an epigenetic phenomenon leading to monoallelic expression of genes based on their parental origin [14]. This process is regulated by epigenetic marks, primarily DNA methylation, which are established in a parent-of-origin-specific manner during gametogenesis and maintained throughout development.
Imprinted genes are vital for prenatal growth, placental development, and postnatal physiology. Disruption of their expression is linked to numerous human diseases, including Prader-Willi syndrome, Angelman syndrome, and Beckwith-Wiedemann syndrome, as well as more common conditions like obesity, diabetes, and psychiatric disorders [14] [15]. The Infinium MethylationEPIC v2 BeadChip provides extensive coverage of known imprinted regions, allowing researchers to investigate perturbations in sperm that may have consequences for offspring health.
Environmental Response: Sperm Epigenetics as a Sensor
The physiological systems of organisms, including humans, act as an interface between environmental change and biological function [16]. Spermatogenesis is highly sensitive to environmental fluctuations, including temperature, toxins, and nutrition [11] [16]. These exposures can induce epigenetic changes in sperm, altering DNA methylation patterns at genes critical for development and health [2] [16].
Emerging evidence suggests that sperm epigenetics serves as a record of paternal environmental exposures. Studies have linked air pollution, endocrine disruptors, and other toxins to altered sperm DNA methylation, which may in turn be associated with adverse offspring birth outcomes and disease susceptibility later in life [2]. Therefore, profiling sperm methylation with the EPICv2 array offers a powerful tool for identifying biomarkers of environmental exposure and understanding their biological consequences.
The Infinium MethylationEPIC v2 BeadChip in Sperm Epigenetics
The Infinium MethylationEPIC v2 BeadChip (EPICv2) is the latest generation of Illumina's methylation arrays, featuring 937,690 probes for interrogating DNA cytosine modifications [4]. Its design offers significant advantages for sperm epigenetics research.
Table: Comparison of Infinium Methylation BeadChip Arrays
| Feature | HM450 | EPICv1 | EPICv2 |
|---|---|---|---|
| Total Probes | 486,427 | 866,552 | 937,690 |
| CpG Loci (cg probes) | ~480,000 | ~865,000 | ~930,000 |
| Coverage of Enhancers | Limited | Expanded | Further Improved |
| Probe Mapping to GRCh38 | Good | Some issues | Best |
| Influence by Genetic Variation | Present | Present | Reduced |
| Low-Input DNA Support | -- | -- | Down to 1 ng |
| Somatic Mutation Probes (nv) | No | No | 824 probes |
Key Features for Sperm Research:
- Comprehensive Coverage: EPICv2 expands coverage on enhancer regions and other regulatory elements, providing a more complete picture of the sperm epigenome [4].
- Improved Design: It features superior probe mapping to the GRCh38 reference genome and reduced susceptibility to confounding by population-specific genetic variation, enhancing data quality and reliability in diverse cohorts [4].
- Low-Input DNA Application: The platform is validated for use with DNA inputs as low as one nanogram, a critical feature for working with limited sperm samples [4].
- Reproducibility: Technical replicates on the EPICv2 platform show high correlation (Spearman's rho > 0.99), ensuring robust and reliable data generation [4].
Essential Protocols for Sperm Epigenetic Analysis
Protocol: Tackling Somatic DNA Contamination in Sperm Studies
Somatic cell contamination in semen samples is a major confounder in sperm epigenetics, as it introduces a distinct methylation signature. The following comprehensive protocol is essential for drawing error-free conclusions [2].
Title: Workflow to Eliminate Somatic DNA Contamination
Detailed Methodology:
- Initial Quality Check: Microscopic Examination
- Examine a fresh aliquot of the semen sample under a microscope to visually assess the presence of round cells (e.g., leukocytes, immature germ cells) alongside mature, flagellated sperm.
Somatic Cell Lysis:
- Treat the semen sample with a Somatic Cell Lysis Buffer (SCLB). This buffer typically contains detergents that selectively lyse the membranes of round somatic cells while leaving sperm cells intact, as their nuclei are protected by resistant membranes and protamines.
- Following lysis, centrifuge the sample to pellet the intact sperm cells and remove the lysate containing somatic cell DNA.
DNA Processing and Interrogation:
- Extract DNA from the purified sperm pellet.
- Proceed with standard bisulfite conversion and processing on the EPICv2 BeadChip as per manufacturer's protocols.
Bioinformatic Quality Control:
- Utilize a published panel of 9,564 CpG sites that are highly methylated in blood cells but unmethylated in pure sperm. These serve as biomarkers for contamination [2].
- Calculate the median methylation beta value across these biomarker CpGs for each sample.
- Apply a 15% cut-off: Samples with a median methylation value of ≥15% at these sites indicate significant somatic contamination and should be excluded from downstream analysis [2].
Protocol: Interrogating Sperm-Specific Epigenetic Programming
This protocol outlines an experimental design to investigate the functional role of sperm epigenetic marks, inspired by studies in model organisms [13].
Research Reagent Solutions:
- Infinium MethylationEPIC v2 BeadChip: For genome-wide DNA methylation profiling of sperm and control cells.
- Somatic Cell Lysis Buffer (SCLB): For purification of sperm cells from semen or testicular tissue.
- Antibodies for Histone Modifications (e.g., H3K4me3, H3K27me3): For Chromatin Immunoprecipitation (ChIP) assays to validate array findings.
- Bisulfite Conversion Kit: For preparing DNA for methylation analysis on the EPICv2 platform.
- DNMT and SETD2 Inhibitors/Model Cell Lines: For functional perturbation studies to establish causality between epigenetic marks and gene regulation [4].
Experimental Workflow:
- Sample Generation:
- Generate embryos using sperm and, for comparison, spermatids (their immediate precursors) via Intracytoplasmic Sperm Injection (ICSI). In model systems, sperm-derived embryos develop more successfully, partly due to superior epigenetic programming [13].
- Epigenetic Profiling:
- Ispure DNA from mature sperm and spermatids.
- Perform DNA methylation analysis using the EPICv2 array.
- Transcriptomic Analysis:
- Collect haploid, paternally-derived embryos from both sperm and spermatid groups at the gastrula stage.
- Perform RNA-seq to identify genes that are misregulated in spermatid-derived embryos.
- Data Integration:
- Correlate differential DNA methylation patterns between sperm and spermatids with the misregulated genes in the resulting embryos. This identifies candidate epigenetic marks delivered by sperm that are essential for correct embryonic gene regulation [13].
- Functional Validation:
- Experimentally perturb identified epigenetic marks (e.g., using inhibitors or in mutant models) in sperm and assess the resulting impact on embryonic gene expression and development to confirm their functional role [13].
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Materials for Sperm Epigenetics Research
| Item | Function/Application |
|---|---|
| Infinium MethylationEPIC v2 BeadChip | Genome-wide profiling of DNA methylation in human sperm; offers enhanced coverage of regulatory elements and supports low-input DNA [4]. |
| Somatic Cell Lysis Buffer (SCLB) | Selectively lyses contaminating round somatic cells in semen samples prior to DNA extraction, crucial for obtaining pure sperm epigenomic data [2]. |
| Bisulfite Conversion Kit | Converts unmethylated cytosines to uracils while leaving methylated cytosines intact, enabling methylation detection on the BeadChip platform. |
| Panel of 9,564 CpG Biomarkers | Bioinformatic tool to quantify and screen out samples with significant somatic cell contamination post-array processing [2]. |
| Antibodies for Histone Modifications | Validate histone retention and modification patterns in sperm via ChIP-seq/qPCR, complementing the DNA methylation data from the array [13]. |
| DNMT/SETD2 Loss-of-Function Models | Cell line models with disruptions in epigenetic writers (e.g., DNMTs, SETD2) to study the mechanistic links between somatic mutations, the epigenetic landscape, and sperm function [4]. |
The paternal germline epigenome is increasingly recognized as a potential contributor to offspring health and development, including neurodevelopmental outcomes. Growing evidence suggests that epigenetic marks in sperm, particularly DNA methylation (DNAm), can reflect paternal exposures and genetic makeup and may be associated with the risk of neurodevelopmental conditions such as autism spectrum disorder (ASD) in offspring [17] [18]. This application note synthesizes evidence from key human cohort studies that have investigated associations between sperm differentially methylated regions (DMRs) and child neurodevelopmental traits, framing these findings within the practical context of utilizing Infinium Methylation BeadChip technology for sperm epigenetics research. We provide detailed protocols, data analysis frameworks, and technical considerations to guide researchers in this emerging field.
Key Findings from Human Cohort Studies
Paternal Sperm DMRs and Child Autistic Traits
The Early Autism Risk Longitudinal Investigation (EARLI), a pregnancy cohort that enrolls mothers who already have a child with ASD, has provided prospective evidence linking the paternal sperm epigenome to child neurodevelopment. In this cohort, genome-scale sperm DNA methylation was measured using the Comprehensive High-throughput Arrays for Relative Methylation (CHARM) array, and autistic traits in children at 36 months were assessed using the Social Responsiveness Scale (SRS) [17].
Table 1: Key Findings from the EARLI Cohort Study on Sperm DMRs and Child Autistic Traits
| Analysis Focus | Number of Significant DMRs Identified | Statistical Threshold | Key Annotations/Overlaps |
|---|---|---|---|
| Child SRS-associated DMRs | 94 | FWER p < 0.05 | Genes implicated in ASD and neurodevelopment |
| Paternal SRS-associated DMRs | 14 | FWER p < 0.05 | - |
| Overlapping DMRs (paternal and child SRS) | 6 | FWER p < 0.1 | - |
| Overlap with previous infant (12-month) autistic trait findings | 16 | FWER p < 0.05 | - |
| Overlap with postmortem brain ASD DMRs | Present (number not specified) | - | CpG sites in child SRS-associated DMRs |
This study demonstrates that paternal germline methylation is associated with autistic traits in 3-year-old offspring, highlighting sperm epigenetic mechanisms as a potential pathway in autism etiology [17]. The findings underscore the utility of epigenome-wide association studies (EWAS) in sperm for identifying potential risk markers for neurodevelopmental outcomes.
Genetic Susceptibility and Neonatal DNA Methylation
Complementing the work on direct sperm epigenetics, other studies have investigated how genetic susceptibility to neurodevelopmental conditions manifests in epigenetic markers at birth. A large meta-analysis of cord blood DNAm from 5,802 participants in four population-based North European cohorts explored associations with polygenic scores (PGS) for ASD, attention-deficit/hyperactivity disorder (ADHD), and schizophrenia (SCZ) [19].
Table 2: Cord Blood DNAm Associations with Polygenic Scores for Neurodevelopmental Conditions
| Polygenic Score (PGS) | Probe-Level Significant Loci | Regional Analysis (DMRs) | Top Findings/Characteristics |
|---|---|---|---|
| SCZ-PGS | 246 loci (p < 9×10⁻⁸) | 157 DMRs | Strong enrichment in Major Histocompatibility Complex; immune-related pathways |
| ASD-PGS | 8 loci | 130 DMRs | Mapped to FDFT1 and MFHAS1 |
| ADHD-PGS | None identified | 166 DMRs | - |
The study found that DNAm signals showed little overlap between the different PGSs, suggesting largely distinct epigenetic correlates of genetic susceptibility across neurodevelopmental conditions [19]. This supports an early-origins perspective for these conditions and indicates that cord blood DNAm may capture congenital biological changes related to genetic risk.
Experimental Protocols & Workflows
Sperm Collection, Processing, and DNA Extraction
Proper handling and processing of sperm samples is critical for obtaining high-quality, contamination-free DNA for methylation studies.
Protocol Details:
- Initial Processing: Fresh semen samples are washed twice with 1X PBS by centrifugation at 200 × g for 15 minutes at 4°C [20].
- Quality Assessment: The washed sample is inspected under a microscope (e.g., 20X objective) to identify the level of somatic cell contamination and perform a sperm count [20].
- Somatic Cell Lysis:
- Incubate sample with freshly prepared Somatic Cell Lysis Buffer (SCLB) (0.1% SDS, 0.5% Triton X-100 in ddH₂O) for 30 minutes at 4°C [20].
- Re-examine under a microscope to detect remaining somatic cells.
- If somatic cells are detected, pellet sperm by centrifugation and repeat SCLB treatment until no somatic cells are visible.
- DNA Extraction:
- After final SCLB treatment and confirmation of somatic cell removal, pellet sperm by centrifugation.
- Perform final PBS wash to obtain a highly pure sperm population [20].
- Extract genomic DNA using automated systems such as the QIAgen QIAsymphony with the Blood 1000 protocol of the DSP DNA Midi kit, following manufacturer's instructions [17].
Addressing Somatic DNA Contamination in Sperm Studies
Semen samples, particularly from oligozoospermic individuals, are often contaminated with somatic cells whose different methylome can bias results [20]. A comprehensive approach is recommended:
- Microscopic Examination: Initial visual inspection to identify gross somatic cell contamination.
- Somatic Cell Lysis Buffer (SCLB) Treatment: Chemical treatment to lyse somatic cells as described in Section 3.1.
- Biomarker Assessment: Utilize CpG sites that are highly methylated in somatic cells but hypomethylated in sperm as contamination markers. By comparing 450K array data between sperm and blood, 9,564 CpG sites with >80% methylation in blood and <20% methylation in sperm (unrelated to infertility) have been identified as potential biomarkers [20].
- Analytical Cut-off: Apply a 15% cut-off during data analysis to eliminate the influence of residual somatic contamination [20].
DNA Methylation Measurement Using Infinium BeadChip Arrays
The Infinium Methylation BeadChip platform is widely used for epigenome-wide DNA methylation analysis due to its cost-effectiveness, quantitative accuracy, and user-friendly data analysis [4].
Platform Options:
- Infinium HumanMethylation450K BeadChip: Covers >485,000 methylation sites including CpG islands, shores, shelves, gene promoters, and enhancers [21].
- Infinium MethylationEPIC (EPIC) BeadChip: Expanded coverage to >850,000 sites with improved enrichment of enhancer regions [4].
- Infinium MethylationEPIC v2.0 BeadChip: Latest version covering ~935,000 sites with improved probe mapping and utility across diverse populations [4].
Methylation Measurement Workflow:
Protocol Details:
- DNA Treatment: Treat DNA (1-4 μg) using bisulfite conversion kits (e.g., 96-well EZ DNA methylation kit) to convert unmethylated cytosines to uracils [17] [21].
- Array Processing: Process converted DNA according to the Infinium HD Methylation assay protocol:
- Scanning: Scan arrays using the iScan System [4].
- Data Extraction: Process raw data using software such as GenomeStudio Methylation Module or open-source tools like SeSAMe in R/Bioconductor to obtain β-values (methylation levels ranging from 0-1) [6] [4].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents and Platforms for Sperm Methylation Studies
| Product/Reagent | Primary Function | Key Features & Applications |
|---|---|---|
| Infinium MethylationEPIC v2.0 BeadChip | Genome-wide DNA methylation profiling | ~935,000 CpG sites; enhanced coverage of enhancers, gene bodies, promoters; suitable for diverse human populations [4] |
| Somatic Cell Lysis Buffer (SCLB) | Removal of somatic cells from semen samples | 0.1% SDS, 0.5% Triton X-100; critical for purifying sperm population for epigenetic analysis [20] |
| QIAsymphony DSP DNA Midi Kit | Automated DNA extraction from sperm | Blood 1000 protocol; high-quality DNA extraction from complex samples [17] |
| EZ DNA Methylation Kit | Bisulfite conversion of DNA | Efficient conversion of unmethylated cytosine to uracil while preserving methylated cytosines [21] |
| CHARM (Custom Array) | Genome-scale methylation analysis | Custom array platform used in EARLI study; covers promoters, miRNA sites, and other genomic features [17] |
Data Analysis Framework
Quality Control and Preprocessing
Robust preprocessing is essential for reliable methylation data:
- Background Correction & Normalization: Use methods like
preprocessNoob()from minfi R package for within-sample normalization and background correction [21]. - Probe Filtering: Remove poorly performing probes with detection p-value > 0.01 in >10% of samples; exclude cross-reactive probes and those containing SNPs [21].
- Batch Effect Correction: Account for technical variation using methods such as ComBat or including batch as a covariate in models [22].
Statistical Analysis for DMR Identification
- Differential Methylation Analysis: Conduct using R packages such as
minfi,DMRcate, orbumphunter[17] [21]. - Covariate Adjustment: Include potential confounders such as genetic ancestry principal components, child sex, age, and batch effects in regression models [17].
- Multiple Testing Correction: Apply family-wise error rate (FWER) or false discovery rate (FDR) control to account for genome-wide testing [17] [21].
Validation and Functional Interpretation
- Independent Replication: Validate findings in independent cohorts when possible.
- Comparison with Public Datasets: Compare identified DMRs with methylation patterns in relevant tissues such as postmortem brains from individuals with ASD [17].
- Functional Annotation: Use pathway analysis tools (e.g., GO, KEGG) to identify biological processes enriched among genes near significant DMRs [17] [21].
Evidence from human cohorts demonstrates that sperm DMRs are associated with child neurodevelopmental outcomes, particularly autistic traits. The Infinium Methylation BeadChip platform provides a robust and cost-effective tool for conducting EWAS in sperm samples, with evolving arrays offering improved genomic coverage and population applicability. However, careful attention to somatic cell contamination, appropriate bioinformatic processing, and validation of findings is essential for generating reliable results. This emerging field holds promise for identifying paternal epigenetic biomarkers of neurodevelopmental risk and understanding intergenerational transmission of disease susceptibility.
The Infinium Methylation BeadChip platform from Illumina has served as the cornerstone technology for large-scale epigenome-wide association studies (EWAS) for over a decade. These arrays have enabled researchers to quantitatively measure DNA methylation levels at cytosine-guanine (CpG) dinucleotides across the human genome, providing critical insights into epigenetic regulation in development, disease, and environmental exposure. The technology operates on the principle of sodium bisulfite conversion, where unmethylated cytosines are converted to uracils while methylated cytosines remain unchanged, allowing for single-base resolution quantification of methylation status through fluorescent detection.
The evolution of this platform has been marked by strategic expansions in genomic coverage, reflecting growing understanding of DNA methylation biology. Each successive array version has incorporated content informed by emerging research, transitioning from a primary focus on promoter-associated CpG islands to encompassing gene bodies, enhancer regions, and other functionally significant genomic elements. This progression has culminated in the latest Infinium MethylationEPIC v2.0 BeadChip (EPICv2), which represents the most comprehensive and technically advanced array to date, offering enhanced capabilities particularly relevant for sperm epigenetics research where unique methylation patterns distinct from somatic tissues are observed [4] [23].
Technical Evolution and Probe Design Advancements
The Infinium Methylation BeadChip platform has undergone substantial evolution since its inception, with each generation expanding genomic coverage and refining technical capabilities:
Table 1: Generational Comparison of Infinium Methylation BeadChips
| Array Characteristic | HumanMethylation450 (450K) | MethylationEPIC v1 (EPICv1) | MethylationEPIC v2 (EPICv2) |
|---|---|---|---|
| Release Year | 2011 | 2016 | 2023 |
| Total Probe Count | 485,577 | 866,552 | ~937,690 |
| CpG Probe Count | ~485,000 | ~865,000 | ~930,000 |
| Retention of 450K Probes | - | ~90% | 81% |
| Retention of EPICv1 Probes | - | - | 83% |
| Infinium I Probe Proportion | ~27% | ~25% | ~23% |
| Key Genomic Coverage | Promoters, CpG islands, gene regions | EPICv1 content + FANTOM5 enhancers | Enhanced coverage of enhancers, CTCF sites, cancer regions |
| Sample Throughput | 12 samples/array | 8 samples/array | 8 samples/array |
| Input DNA Recommendation | 500 ng | 250 ng | 250 ng (validated down to 1 ng) |
The progression from 450K to EPICv1 represented a near-doubling of probe content, with significant expansion into enhancer regions identified by the FANTOM5 project [4] [24]. EPICv2 builds upon this foundation with additional improvements, including the reintroduction of 24,463 cg probes from HM450 that were not present in EPICv1, plus 183,435 completely new cg probes representing 20% of the total EPICv2 content [4]. This strategic selection of new content provides improved coverage of biologically significant regions, including super-enhancers, CTCF-binding sites, and open chromatin regions associated with primary tumors identified by ATAC-Seq and ChIP-seq experiments [25].
Probe Design and Chemistry Refinements
The Infinium assay employs two distinct probe chemistries, both retained across array generations but with proportional adjustments:
- Infinium I Probes: Utilize two separate bead types for methylated and unmethylated states, providing more robust measurement but requiring greater physical space on the array.
- Infinium II Probes: Employ a single bead type with color discrimination between methylation states, allowing higher density profiling.
EPICv2 maintains a similar ratio of Infinium I and II probes as its predecessors, with only minimal changes: 70 Infinium I probes switched to Infinium II chemistry, and 12 Infinium II probes switched to Infinium I [4]. This consistency in chemistry supports data comparability across array versions. A significant advancement in EPICv2 is the improved probe mappability, with fewer probes exhibiting poor mapping to the GRCh38 reference genome and reduced susceptibility to ancestry-specific genetic variation [4]. Of the probes deleted in EPICv2, 72.9% had issues with cross-reactivity or direct influence from sequence polymorphisms, compared to only 0.1% of retained probes, indicating more stringent probe selection criteria [4].
Table 2: Technical Performance Metrics Across Array Generations
| Performance Metric | 450K | EPICv1 | EPICv2 |
|---|---|---|---|
| Technical Reproducibility (Correlation) | >0.99* | >0.99* | >0.99 [4] |
| Cross-hybridizing Probes | ~5.5% [24] | ~5.5% [24] | Reduced but present [24] |
| Probe Mapping Issues | Significant [24] | Significant [24] | Improved [4] |
| Data Concordance with WGBS | High* | High* | High [24] |
| Compatibility with FFPE Samples | Limited | Yes | Yes with modified protocol [25] |
*Based on historical performance data; not directly assessed in current search results
Application in Sperm Epigenetics Research
Sperm-Specific Methodological Considerations
Sperm epigenetics research presents unique challenges due to the distinctive architecture of sperm chromatin, characterized by protamine-bound DNA with retained histones at specific regulatory regions [23]. This unique composition necessitates specialized protocols for sperm cell isolation and DNA extraction to ensure high-quality methylation data. The following protocol has been optimized for sperm epigenetics studies using Infinium BeadChips:
Protocol 1: Sperm Processing for Methylation Analysis
Sperm Isolation
- Layer fresh semen sample onto density gradient (e.g., Isolate, Irvine Scientific)
- Centrifuge at 300-500 × g for 20 minutes
- Collect motile sperm fraction from the bottom
- Wash twice with phosphate-buffered saline (PBS)
Somatic Cell Elimination
- Resuspend sperm pellet in somatic cell lysis buffer (0.1% SDS, 0.5% Triton X-100)
- Incubate on ice for 30 minutes with occasional vortexing
- Centrifuge to collect intact sperm cells
- Confirm absence of somatic contamination by microscopy
DNA Extraction
- Extract genomic DNA using modified guanidinium thiocyanate method or commercial kits
- Quantify DNA using fluorometric methods
- Assess DNA purity (A260/280 ratio ~1.8-2.0)
Bisulfite Conversion
- Use 500-1000 ng sperm DNA for bisulfite conversion
- Employ commercial bisulfite conversion kits (e.g., Zymo EZDNA Methylation Kit)
- Follow manufacturer's protocol with extended conversion time (16-20 hours)
- Purify converted DNA and elute in low TE buffer
Array Processing
- Process according to Illumina Infinium HD Methylation Assay protocol
- Use 250 ng bisulfite-converted DNA per array
- Amplify, fragment, hybridize, and stain according to manufacturer specifications
- Scan arrays using iScan or NextSeq 550 System
This protocol has been successfully applied in multiple sperm epigenetics studies that identified age-associated methylation changes using Infinium arrays [26] [27] [28].
Key Insights from Sperm Methylation Studies
Research utilizing Infinium arrays has revealed fundamental aspects of sperm epigenetics, particularly regarding age-associated methylation changes:
Figure 1: Paternal Age Effect Pathways. Age-related sperm methylation changes preferentially affect genes involved in development and neurodevelopment, potentially influencing offspring outcomes [27] [29].
Studies using 450K and EPIC arrays have consistently demonstrated that advanced paternal age is associated with specific methylation alterations in sperm. A comprehensive analysis of 73 sperm samples using reduced representation bisulfite sequencing (RRBS) identified 1,565 age-associated differentially methylated regions (ageDMRs), with a strong bias toward hypomethylation (74% of ageDMRs) rather than hypermethylation (26%) [27] [29]. These ageDMRs show distinct genomic distributions: hypomethylated regions are predominantly located near transcription start sites, while hypermethylated regions are more frequently found in gene-distal regions [27].
The functional enrichment of genes associated with sperm ageDMRs is particularly notable. Among 241 genes replicated across multiple studies, significant enrichments have been identified in 41 biological processes associated with development and the nervous system, and 10 cellular components associated with synapses and neurons [27] [29]. This pattern supports the hypothesis that paternal age effects on the sperm methylome may contribute to the increased risk of neurodevelopmental disorders in children of older fathers [27] [23].
Experimental Protocols for Cross-Platform Validation
Protocol for Longitudinal Study Design
Integrating data across different array generations is essential for longitudinal studies and meta-analyses. The following protocol enables robust cross-platform validation:
Protocol 2: Cross-Platform Array Comparison
Sample Selection and Design
- Select 30+ samples representing study population
- Include balanced sex representation (15 male, 15 female)
- Ensure coverage of relevant age range and phenotypes
- Include technical replicates (6 for 450K, 2 for EPIC arrays)
Parallel Processing
- Divide each sample for processing on 450K, EPICv1, and EPICv2 arrays
- Use identical bisulfite-converted DNA aliquots
- Process simultaneously using same reagent lots
- Maintain consistent laboratory conditions
Data Processing and Normalization
Quality Assessment Metrics
- Calculate intraclass correlations for shared probes
- Assess interquartile ranges across arrays
- Determine array bias (variance explained by array type)
- Evaluate replicate concordance
Probe Filtering Strategy
- Retain 369,639 CpGs present across all three arrays
- Remove probes with poor performance in any platform
- Annotate probe quality metrics across arrays
- Create platform-harmonized dataset
This approach was successfully implemented in the Drakenstein Child Health Study, which directly compared all three array versions in the same participants [30].
Protocol for Sperm-Specific Epigenetic Clock Validation
Epigenetic clocks based on Infinium array data have emerged as powerful tools for biological age estimation. Their application to sperm requires specific validation:
Protocol 3: Sperm Epigenetic Clock Assessment
Sample Collection and Processing
- Collect sperm samples from men across adult age span (20-60 years)
- Process using Protocol 1 for sperm isolation and DNA extraction
- Perform bisulfite conversion
- Run on EPICv2 arrays following standard protocol
Data Preprocessing
Clock Calculation
- Extract beta values for clock-specific CpG sites
- Apply published coefficients for relevant clocks:
- Horvath pan-tissue clock
- Hannum blood clock
- Skin & blood clock
- GrimAge mortality predictor
- Calculate DNAm age using respective algorithms
Validation Metrics
- Correlate DNAm age with chronological age
- Assess mean absolute error (MAE)
- Evaluate precision (R² value)
- Compare performance across array versions
Studies have shown that principal component-based epigenetic clocks demonstrate greater stability across array versions compared to non-PC-based clocks, with mean absolute percentage errors (MAPE) of 0.118-8.98% versus 5.31-21.2%, respectively [31].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for Sperm Methylation Analysis
| Reagent/Kit | Manufacturer | Function | Sperm-Specific Considerations |
|---|---|---|---|
| Isolate Sperm Separation Medium | Irvine Scientific | Density gradient isolation of motile sperm | Eliminates seminal plasma and immotile sperm |
| EpiTect Bisulfite Kit | Qiagen | Sodium bisulfite conversion of unmethylated cytosines | Extended incubation may improve conversion efficiency |
| Infinium MethylationEPIC v2.0 Kit | Illumina | Genome-wide methylation profiling | Compatible with sperm DNA; 250 ng input recommended |
| Zymo EZDNA Methylation Kit | Zymo Research | Bisulfite conversion alternative | Validated for low-input samples (down to 1 ng) |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Fluorometric DNA quantification | More accurate for sperm DNA than spectrophotometry |
| PyroMark PCR Kit | Qiagen | Amplification for bisulfite pyrosequencing | Enables validation of array findings at specific loci |
The evolution from HM450 to EPICv2 represents significant advancements in probe design, genomic coverage, and technical performance that directly benefit sperm epigenetics research. EPICv2's expanded content in enhancer regions and CTCF-binding sites, combined with improved probe mappability and reduced cross-hybridization, provides enhanced capacity to detect biologically significant methylation alterations in sperm [4] [24]. The platform's backward compatibility with previous arrays (retaining 83% of EPICv1 and 81% of HM450 probes) enables valuable longitudinal analyses and meta-analyses, though careful normalization and probe selection are essential [30] [4].
For sperm epigenetics specifically, the application of EPICv2 promises to advance understanding of paternal age effects, environmental impacts on sperm methylation, and potential transgenerational epigenetic inheritance [23]. The continued development of sperm-specific epigenetic clocks and validation of their biological significance will be crucial for translating array-based findings into clinical applications. As the field progresses, integration of methylation array data with other epigenetic marks, including sperm histones and non-coding RNAs, will provide more comprehensive understanding of how paternal epigenetic information influences embryonic development and offspring health [23].
From Sample to Data: A Best-Practice Workflow for Sperm Methylation Analysis
Sample Collection and DNA Extraction Protocols for Semen Specimens
Sperm epigenetics, particularly DNA methylation, is a critical field of study for understanding male fertility, embryonic development, and transgenerational inheritance [2] [32]. The Infinium Methylation BeadChip has emerged as a predominant technology for profiling genome-wide DNA methylation in sperm due to its cost-effectiveness, quantitative accuracy, and user-friendly data analysis pipelines [4]. However, the unique biological characteristics of spermatozoa present distinct challenges for DNA methylation analysis. The sperm nucleus is characterized by extremely compact chromatin, where histones are replaced by protamines, creating a physical barrier to DNA extraction [33]. Furthermore, semen is a complex fluid containing cellular debris, leucocytes, bacteria, and seminal plasma, all of which can contaminate or interfere with downstream epigenetic analyses [34] [2]. Therefore, rigorous and standardized protocols for semen collection and DNA extraction are fundamental prerequisites for generating high-quality, reproducible DNA methylation data using the Infinium platform. This application note provides detailed methodologies to support researchers in this critical preparatory phase.
Semen Sample Collection and Initial Processing
Proper collection and initial processing are crucial for preserving the integrity of sperm DNA for subsequent epigenetic analysis.
Collection Protocol
- Abstinence Period: A period of 3–7 days is required prior to sample collection [35].
- Collection Method: Samples are collected by masturbation into a sterile plastic container. The use of condoms or lubricants is prohibited, as they may contain spermicidal agents or compounds that interfere with analysis [35].
- Liquefaction: After collection, the semen sample must be allowed to liquefy for 45–60 minutes at room temperature in a dedicated collection room [35].
Initial Semen Analysis and Sperm Separation
Basic semen analysis should be performed according to World Health Organization criteria, measuring volume, pH, concentration, total sperm count, and motility [35]. For DNA extraction aimed at epigenetic studies, it is essential to separate spermatozoa from somatic cells (e.g., leukocytes) present in the ejaculate, as their DNA methylation signatures are distinct and can confound results [2].
Efficient sperm separation techniques, such as Discontinuous Density Gradient Centrifugation (DGC) or swim-up, are recommended. DGC is particularly effective as it selects for morphologically normal, motile spermatozoa and helps remove seminal plasma, non-gametic cells, and other contaminants [34]. The resulting purified sperm pellet is then used for DNA extraction.
Table 1: Standardized Semen Collection Parameters
| Parameter | Specification | Purpose/Rationale |
|---|---|---|
| Abstinence Period | 3–7 days | Ensures optimal sample volume and concentration [35] |
| Collection Method | Masturbation into sterile container | Prevents contamination from external sources |
| Prohibited Materials | Condoms, lubricants | Avoids introduction of spermicides or DNA-inhibiting chemicals [35] |
| Liquefication Time | 45–60 minutes | Allows semen to reach a viscous state suitable for processing [35] |
| Sperm Separation | Density Gradient Centrifugation (DGC) | Isolates sperm from somatic cells and seminal plasma [34] [2] |
The following workflow outlines the journey from semen collection to DNA application, highlighting key quality control checkpoints.
DNA Extraction from Spermatozoa
The compact nature of sperm chromatin, stabilized by disulfide bridges between protamines, necessitates specialized DNA extraction methods that incorporate robust lysis conditions [33].
Optimized In-House DNA Extraction Protocol
This protocol, adapted from comparative methodological studies, uses a combination of reducing agents to effectively break down the sperm's nuclear membrane [33].
Reagents:
- Lysis Buffer: 100 mM Tris-HCl (pH 8.0), 500 mM NaCl, 10 mM EDTA, 1% SDS.
- Reducing Agents: Dithiothreitol (DTT) and β-Mercaptoethanol (β-ME). Note: DTT must be prepared fresh.
- Enzymes: Proteinase K and RNase A.
- Other: Absolute ethanol, 70% ethanol, Triton-X100.
Step-by-Step Procedure:
- Lysis: Mix the purified sperm pellet with lysis buffer. Add both DTT (to a final concentration of 25mM) and β-ME (to a final concentration of 2.5%) [33].
- Incubation: Incubate the mixture at 65°C for 2 hours in a water bath to ensure complete lysis and decondensation of chromatin.
- Enzymatic Treatment:
- Add Proteinase K (200 µg/mL) and continue incubation at 65°C for 1 hour to digest proteins.
- Add RNase A (20 µg/mL) and incubate at 37°C for 30–60 minutes to remove RNA contamination.
- DNA Precipitation: Add an equal volume of absolute ethanol to the lysate to precipitate the DNA. Gently invert the tube until DNA threads become visible.
- Washing: Centrifuge to pellet the DNA. Wash the pellet thoroughly with 70% ethanol to remove salts and other impurities.
- Rehydration: Air-dry the DNA pellet and reconstitute it in a suitable buffer (e.g., TE buffer or nuclease-free water).
Comparison of DNA Extraction Methods
A systematic comparison of extraction methods for caprine sperm, relevant to human sperm studies, evaluated protocols based on DNA yield, purity (A260/280 ratio), and integrity. The results are summarized below.
Table 2: Functional Comparison of DNA Extraction Methods from Sperm [33]
| Extraction Method | Key Characteristic | Average DNA Yield (Fresh Sperm) | Average A260/280 Ratio | Suitability for Sequencing |
|---|---|---|---|---|
| In-House (DTT + β-ME) | Combination of reducing agents | ~1250 ng/µL | ~1.85 | Excellent |
| Commercial Kit A | Silica-column based | ~850 ng/µL | ~1.75 | Good |
| Phenol-Chloroform | Organic solvent extraction | ~650 ng/µL | ~1.65 | Moderate |
| Protocol with DTT only | Single reducing agent | ~950 ng/µL | ~1.80 | Good |
| Protocol with β-ME only | Single reducing agent | ~750 ng/µL | ~1.78 | Moderate |
The data demonstrates that the in-house method utilizing a combination of DTT and β-ME outperforms other methods, yielding DNA with superior concentration and purity, making it highly suitable for genome-wide studies like Infinium BeadChip analysis [33].
Quality Control for Downstream Methylation Analysis
Prior to proceeding with the Infinium BeadChip, extracted DNA must pass stringent quality control checks.
- Purity and Quantity: Assess DNA concentration using a fluorometer (e.g., Qubit). Measure purity using spectrophotometry (e.g., NanoDrop); an A260/280 ratio between 1.8 and 2.0 indicates minimal protein contamination [33] [36].
- Integrity: Verify DNA integrity using gel electrophoresis, ensuring the presence of a high-molecular-weight band with minimal smearing, which indicates low fragmentation [33].
- Input Requirements: The Infinium MethylationEPIC v2 BeadChip supports DNA inputs as low as 1 nanogram, demonstrating robustness for samples with limited yield [4]. Standard inputs of 500 ng are commonly used for bisulfite conversion prior to array hybridization [36].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Kits for Sperm DNA Methylation Studies
| Item | Function/Application | Specific Example/Note |
|---|---|---|
| Dithiothreitol (DTT) | Reducing agent critical for breaking disulfide bonds in protamine-compacted sperm chromatin [33] | Use fresh preparations; final concentration of 25mM in lysis buffer [33] |
| β-Mercaptoethanol (β-ME) | Reducing agent used in combination with DTT for enhanced sperm cell lysis [33] | Final concentration of 2.5% in lysis buffer [33] |
| Proteinase K | Broad-spectrum serine protease for digesting proteins and nucleases during extraction [33] | Typical working concentration of 200 µg/mL [33] |
| Infinium MethylationEPIC v2 BeadChip | Microarray for genome-wide DNA methylation profiling at > 935,000 CpG sites [4] | Covers enhancer regions and is applicable to diverse ancestry groups [4] |
| Somatic Cell Lysis Buffer | Selective lysis of contaminating somatic cells (e.g., leukocytes) in semen prior to sperm DNA extraction [2] | Critical step to prevent confounding methylation signals from somatic DNA [2] |
| Bisulfite Conversion Kit | Chemical treatment (e.g., EZ DNA Methylation Kit) that converts unmethylated cytosines to uracils for downstream detection on the BeadChip [36] | A mandatory step prior to hybridization on the Infinium BeadChip |
The reliability of sperm DNA methylation data generated using the Infinium Methylation BeadChip is fundamentally dependent on the initial steps of sample collection and preparation. Adherence to a standardized semen collection protocol, followed by efficient sperm separation and a DNA extraction method optimized for sperm's unique chromatin structure—specifically one incorporating potent reducing agents like DTT and β-ME—ensures the isolation of high-quality, contaminant-free genomic DNA. The detailed protocols and comparative data provided herein offer a robust framework for researchers to generate high-fidelity DNA samples, thereby laying a solid foundation for meaningful and reproducible sperm epigenetics research.
Bisulfite Conversion and Low-Input DNA Considerations (Down to 1 ng)
DNA methylation analysis using the Infinium Methylation BeadChip is a cornerstone of modern sperm epigenetics research, enabling investigations into fertility, transgenerational inheritance, and environmental exposures [2] [37]. The process begins with conversion treatment, which creates sequence-based differences between methylated and unmethylated cytosines. For decades, bisulfite conversion (BC) has been the gold standard method, but recent advances in enzymatic conversion (EC) technologies and optimized bisulfite protocols now offer researchers multiple paths forward, especially critical when working with the limited DNA quantities typical of forensic or clinically derived semen samples [38] [39]. The fundamental challenge lies in balancing conversion efficiency with DNA preservation, as the conversion method directly impacts data quality, coverage, and the validity of conclusions drawn about sperm methylation patterns.
This application note provides a structured comparison of conversion methods and detailed protocols tailored for sperm epigenetics research utilizing Infinium Methylation BeadChips, with particular emphasis on handling low-input DNA samples down to 1 ng.
Comparison of DNA Conversion Methods
Performance Metrics for Low-Input DNA
The choice between conversion methods involves trade-offs between DNA recovery, fragmentation, and conversion efficiency. These factors become critically important when working with low-input DNA, such as that obtained from limited sperm samples.
Table 1: Quantitative Performance Comparison of DNA Conversion Methods for Low-Input DNA
| Performance Metric | Conventional Bisulfite (CBS) | Enzymatic Conversion (EC) | Ultra-Mild Bisulfite (UMBS) |
|---|---|---|---|
| Minimum Reliable Input | 5 ng [38] | 10 ng [38] | As low as 10 pg [39] |
| Conversion Efficiency | >99.5% [40] [41] | ~94-99.9% [38] [40] [41] | ~99.9% [39] |
| DNA Recovery Rate | 61-81% (cfDNA) [41], but overestimated in assays [38] | 30-47% (cfDNA) [41], 40% (genomic DNA) [38] | Significantly higher than CBS and EM-seq [39] |
| Fragmentation Level | High (14.4 ± 1.2) [38] | Low-Medium (3.3 ± 0.4) [38] | Significantly reduced vs. CBS [39] |
| Background Noise (Unconverted C) | <0.5% [39] | Can exceed 1% at low inputs [39] | ~0.1% across all inputs [39] |
| Library Complexity | Lower duplication rates [39] | Higher than CBS [39] | Highest; outperforms both CBS and EM-seq [39] |
| Protocol Duration | 12-16 hours incubation [38] | ~6 hours total [38] | ~90 minutes incubation [39] |
| Cost per Reaction | ~€2.91 [38] | ~€6.41 [38] | Information not specified |
Technical Considerations for Sperm Epigenetics
When applying these methods specifically to sperm research, several unique considerations emerge:
- Somatic Cell Contamination: Sperm samples often contain somatic cell contamination that significantly alters methylation profiles. Implement comprehensive quality control including microscopic examination, somatic cell lysis buffer treatment, and analysis of 9,564 established blood-specific CpG markers with a 15% cutoff threshold to eliminate contaminated samples [2].
- Age Prediction Applications: For epigenetic age prediction in semen, targeted approaches have identified optimal marker sets. A 6-CpG model (SH2B2, EXOC3, IFITM2, GALR2, and FOLH1B) predicts age with MAE of 5.1 years, representing a practical balance between accuracy and multiplexing feasibility for forensic applications [37].
- Disease Association Studies: Abnormal methylation patterns in sperm, such as hypermethylation of ST8SIA4 in low-motility sperm, can be detected using the MethylationEPIC array, highlighting the importance of high-quality conversion for biomarker discovery [42].
Method Selection Workflow
Figure 1: Decision workflow for selecting appropriate DNA conversion methods based on input quantity and research requirements for sperm epigenetics studies.
Detailed Experimental Protocols
Ultra-Mild Bisulfite Conversion (UMBS-seq) for Minimal Input
The UMBS-seq protocol represents the cutting edge for low-input sperm methylation studies, enabling work with samples as limited as 10 pg of DNA [39].
Reagents and Equipment:
- Ultra-Mild Bisulfite Reagent: 100 μL of 72% ammonium bisulfite + 1 μL of 20 M KOH
- DNA Protection Buffer (included in commercial kits)
- Thermal cycler with precise temperature control
- Magnetic bead-based purification system (e.g., AMPure XP)
Step-by-Step Procedure:
- DNA Denaturation: Dilute DNA sample to 10 μL with nuclease-free water. Add 2 μL of DNA Protection Buffer. Incubate at 55°C for 20 minutes.
- Ultra-Mild Bisulfite Conversion: Prepare fresh UMBS reagent (100 μL 72% ammonium bisulfite + 1 μL 20 M KOH). Add 52 μL UMBS reagent to denatured DNA. Incubate at 55°C for 90 minutes.
- Desalting and Purification: Use magnetic bead-based cleanup (1.8x bead ratio) to remove bisulfite salts. Elute in 20 μL nuclease-free water.
- Desulfonation: Add 4 μL of 1 M NaOH and incubate at room temperature for 15 minutes.
- Final Purification: Perform additional magnetic bead cleanup (1.8x ratio). Elute in 20 μL nuclease-free water.
- Quality Assessment: Use qBiCo or BisQuE multiplex qPCR to assess conversion efficiency, DNA recovery, and fragmentation before proceeding to BeadChip analysis.
Critical Steps for Success:
- Always prepare UMBS reagent fresh before each conversion
- Use high-quality nuclease-free water to prevent degradation
- Implement rigorous temperature control at 55°C ± 0.5°C
- For inputs below 1 ng, increase magnetic bead ratio to 3.0x to improve recovery
Enzymatic Conversion for Moderate Input DNA
For sperm DNA samples in the 10-200 ng range, enzymatic conversion provides an excellent balance of preservation and efficiency.
Reagents and Equipment:
- NEBNext Enzymatic Methyl-seq Conversion Module
- Magnetic beads (AMPure XP recommended)
- Thermal cycler
- Microcentrifuge
Step-by-Step Procedure:
- DNA Fragmentation (Optional): For intact genomic DNA, fragment to 300 bp using Covaris shearing. For already fragmented sperm DNA, proceed directly.
- Oxidation and Glycosylation: Set up reaction with 10-200 ng DNA in 45 μL water. Add 5 μL TET2 Oxidation Buffer and 1.5 μL TET2 Enzyme. Incubate at 37°C for 1 hour. Add 5 μL T4-BGT Buffer and 1.5 μL T4-BGT Enzyme. Incubate at 37°C for 1 hour.
- First Cleanup: Use AMPure XP beads at 1.8x ratio. Elute in 32 μL nuclease-free water.
- APOBEC Deamination: Add 5 μL APOBEC Buffer and 1.5 μL APOBEC Enzyme to purified DNA. Incubate at 37°C for 1 hour.
- Second Cleanup: Use AMPure XP beads at 1.8x ratio. Elute in 20 μL nuclease-free water.
- Quality Control: Assess conversion using ddPCR with Chr3 and MYOD1 assays [41].
Optimization for Low Recovery:
- If recovery is suboptimal, increase bead-to-sample ratio to 3.0x for both cleanup steps
- Test different magnetic bead brands (AMPure XP, Mag-Bind TotalPure NGS, NEBNext Sample Purification Beads)
- Extend oxidation and glycosylation incubation times to 90 minutes each for <20 ng inputs
Quality Control Assessment Protocols
qBiCo Multiplex qPCR Assessment [38]:
- Prepare qPCR reaction with 5 μL converted DNA, 10 μL 2× qPCR master mix, 0.5 μL each primer/probe mix
- Use the following cycling conditions: 95°C for 10 min, 45 cycles of 95°C for 15 sec and 60°C for 1 min
- Calculate conversion efficiency = 1 - (Genomic/Converted) × 100%
- Determine fragmentation index = (Long amplicon Ct - Short amplicon Ct)
- Compute recovery rate = (Converted DNA quantity / Input DNA quantity) × 100%
BisQuE Alternative Protocol [40]:
- Uses two different-sized multicopy regions (104 bp and 238 bp) with cytosine-free primers
- Includes artificial IPC to check for PCR inhibitors
- Enables simultaneous assessment of conversion efficiency, recovery, and degradation in a single assay
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagent Solutions for Bisulfite Conversion and Methylation Analysis
| Reagent/Category | Specific Examples | Function & Application Notes |
|---|---|---|
| Bisulfite Kits | Zymo EZ DNA Methylation-Lightning, Qiagen EpiTect Fast, UMBS formulation | Chemical conversion of unmethylated C to U; UMBS offers reduced damage for low inputs [39] [40] |
| Enzymatic Kits | NEBNext Enzymatic Methyl-seq Conversion Module | Enzyme-based conversion; gentler on DNA but lower recovery; ideal for moderate inputs [38] [41] |
| Magnetic Beads | AMPure XP, NEBNext Sample Purification Beads, Mag-Bind TotalPure NGS | DNA cleanup and size selection; critical for recovery optimization at low inputs [41] |
| DNA Polymerases | Q5U Hot Start High-Fidelity DNA Polymerase, NEBNext Q5U Master Mix | Amplification of uracil-rich bisulfite-converted DNA; essential for library prep [43] |
| Quality Control | qBiCo, BisQuE, ddPCR with Chr3/MYOD1 assays | Assess conversion efficiency, DNA recovery, and fragmentation before BeadChip [38] [40] [41] |
| Methylation Arrays | Infinium MethylationEPIC v2 BeadChip | Comprehensive methylation profiling; supports inputs down to 1 ng [4] |
| Library Prep | NEBNext Ultra II DNA Library Prep Kit | Compatible with bisulfite-converted DNA; enables sequencing validation [43] |
Successful bisulfite conversion of low-input DNA for sperm epigenetics research requires careful method selection based on DNA quantity and quality requirements. For the most challenging samples with inputs below 1 ng, Ultra-Mild Bisulfite methods currently provide the optimal balance of conversion efficiency and DNA preservation. For standard sperm epigenetics studies with 10-200 ng input, enzymatic conversion offers substantial benefits in DNA integrity, while conventional bisulfite remains cost-effective for higher inputs. By implementing the rigorous quality control measures and optimized protocols outlined herein, researchers can reliably generate high-quality methylation data from precious sperm samples, advancing our understanding of male fertility and epigenetic inheritance.
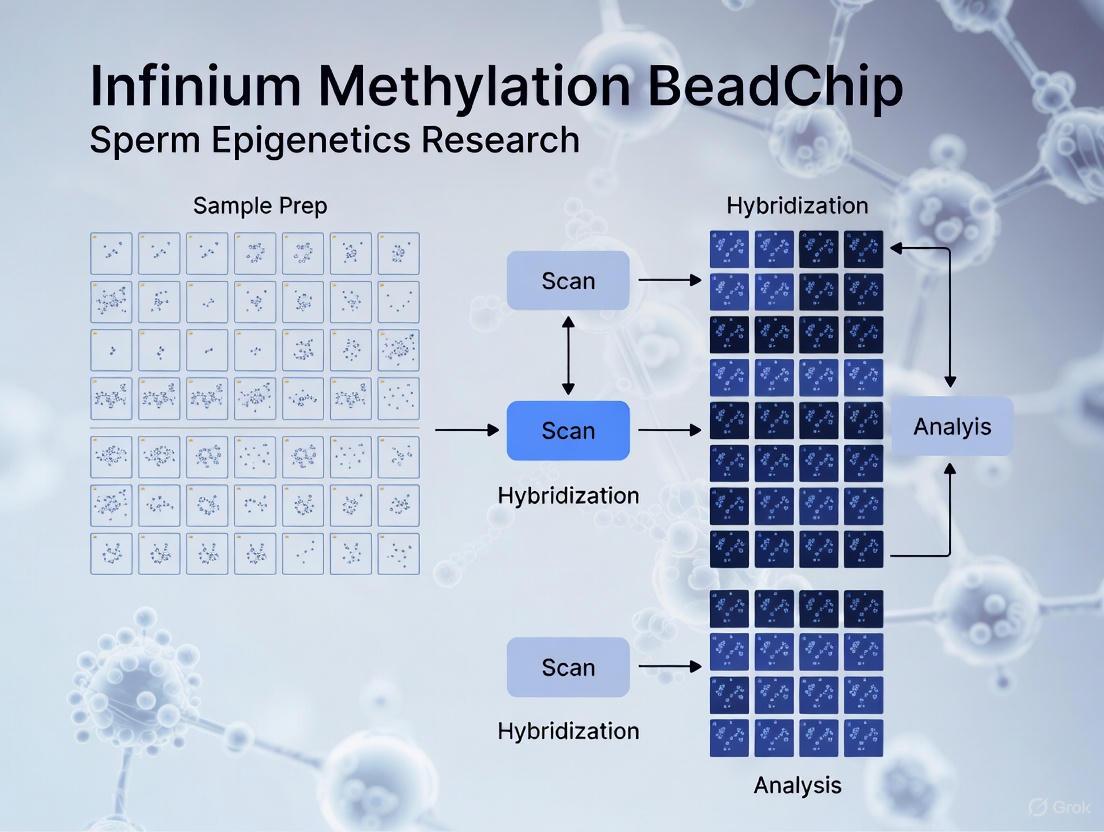
Hybridization, Staining, and Processing on the BeadChip Platform
The Infinium Methylation BeadChip platform provides a high-throughput, cost-effective solution for epigenome-wide association studies (EWAS), enabling robust profiling of DNA methylation status across hundreds of thousands of CpG sites. For sperm epigenetics research, this technology offers a powerful tool to investigate correlations between sperm methylation patterns and factors such as fertility, environmental exposures, and transgenerational inheritance [2]. This application note details the protocols for sample processing, hybridization, staining, and data analysis, with specific considerations for sperm-derived DNA to ensure data integrity and biological relevance.
Principle of the Infinium Methylation Assay
The core of the technology relies on probing the methylation status of CpG sites after sodium bisulfite conversion, which deaminates unmethylated cytosines to uracils while leaving methylated cytosines unchanged [44] [45]. The assay then uses two different probe design chemistries to interrogate the CpG loci:
- Infinium I (Two-Probe Design): Employs two separate probes per CpG locus—one for the methylated state and one for the unmethylated state. The 3' terminus of each probe is complementary to either the cytosine (methylated) or thymine (unmethylated) base. A single-base extension with a fluorescently labeled nucleotide determines the state [6] [45].
- Infinium II (Single-Probe Design): Uses a single probe per CpG locus. The methylation state is determined during the single-base extension step that occurs after hybridization, which incorporates a dye-labeled nucleotide [6].
Following hybridization and extension, the BeadChip is stained and scanned on a system such as the iScan to measure fluorescence intensities. The relative methylation level at each CpG site is calculated as a beta value (β), where β = IntensityMethylated / (IntensityMethylated + Intensity_Unmethylated + 100) [46]. The beta value ranges from 0 (completely unmethylated) to 1 (fully methylated).
Materials and Equipment
Research Reagent Solutions
The following reagents and equipment are essential for performing the Infinium Methylation Assay.
Table 1: Essential Reagents and Equipment for the Infinium Methylation Assay
| Item | Function/Description | Example/Part Number |
|---|---|---|
| EZ-96 DNA Methylation Kit | For bisulfite conversion of genomic DNA. | Zymo Research, D5003 [44] |
| Infinium HD Methylation BeadChip Kit | Contains BeadChips and reagents for amplification, fragmentation, hybridization, labeling, and staining. | Varies by species (e.g., Human MethylationEPIC v2.0) [6] [47] |
| Infinium Methylation Assay Buffers | Includes MSM, FMS, PM1, LMX, ATM, and Staining solutions for the various assay steps. | Included in BeadChip Kit [44] |
| iScan System | Scanner for imaging the fluorescent signals from the processed BeadChips. | Illumina [6] [44] |
| Bisulfite-Converted DNA | The starting material for the assay. Input of 250 ng is recommended, though lower inputs have been tested [6] [46]. | N/A |
Special Considerations for Sperm Epigenetics
Sperm DNA is particularly susceptible to somatic DNA contamination, which can severely skew methylation results. A comprehensive plan to address this is critical [2]:
- Microscopic examination of semen samples.
- Treatment with a somatic cell lysis buffer (SCLB).
- Utilization of 9,564 identified CpG markers that are highly methylated in blood but not in sperm to quantify contamination.
- Application of a 15% cutoff during data analysis to exclude samples with significant somatic contamination [2].
Experimental Protocol
Bisulfite Conversion of Genomic DNA
The initial and most critical wet-lab step is the bisulfite conversion of DNA, which must be performed prior to the BeadChip assay.
- DNA Quantitation and Dilution: Quantitate genomic DNA using a spectrophotometer (e.g., NanoDrop). The A260/280 ratio should be >1.80. Dilute DNA to a concentration of 100 ng/μL in PCR-grade water [44].
- Reagent Preparation: Prepare the CT Conversion Reagent from the EZ-96 DNA Methylation Kit by adding 7.5 mL of water and 2.1 mL of M-Dilution Buffer to the CT Conversion Reagent bottle. Vortex frequently for 10 minutes until mostly dissolved. Prepare the M-Wash Buffer by adding 144 mL of 100% ethanol to the 36 mL of concentrate [44].
- Conversion Reaction: Pipette 10 μL of each DNA sample (100 ng/μL, 1 μg total) into a 96-well plate. Add 5 μL of M-Dilution Buffer and adjust the total volume to 50 μL with water. Seal the plate and incubate at 37°C for 15 minutes. Add 100 μL of the prepared CT Conversion Reagent to each well, mix by pipetting, and seal the plate [44].
- Thermal Cycling: Incubate the plate on a thermal cycler using the following program [44]:
- 95°C for 30 seconds
- 50°C for 60 minutes
- Repeat the above two steps for 15 more cycles.
- 4°C hold.
- DNA Purification and Desulfonation: Transfer the reacted samples to a silica-binding plate placed on a collection plate that has been pre-loaded with 400 μL of M-Binding Buffer. Centrifuge at ≥3,000 x g for 5 minutes. Wash the bound DNA by adding 500 μL of M-Wash Buffer and centrifuging. Add 200 μL of M-Desulphonation Buffer, incubate at room temperature for 15–20 minutes, and centrifuge. Perform two additional washes with 500 μL of M-Wash Buffer, with the final centrifugation for 10 minutes to ensure all ethanol is removed [44].
- Elution: Place the binding plate on a new elution plate. Add 30 μL of M-Elution Buffer directly to the silica matrix in each well, incubate for 1–2 minutes, and centrifuge at ≥3,000 x g for 3 minutes to elute the bisulfite-converted DNA [44].
- Quality Control: Determine the concentration of the bisulfite-converted DNA. Assess the conversion efficiency using quality control methods such as methylation-specific PCR or pyrosequencing of repetitive elements like LINE1 [44]. For sperm DNA, it is highly recommended to also run a contamination check using the somatic CpG markers mentioned in Section 3.2 [2].
BeadChip Processing: Amplification, Hybridization, Staining, and Scanning
The following workflow describes the steps for processing the bisulfite-converted DNA on the BeadChip. The protocol can be performed manually or in an automated workflow using systems like the Infinium Automated Pipetting System (IAPS) [45].
Diagram 1: BeadChip processing workflow
- Whole Genome Amplification (WGA): The bisulfite-converted DNA is isothermally amplified overnight. This step non-specifically amplifies the converted DNA several hundred-fold, replacing uracils with thymines [44] [45].
- Fragmentation: The amplified DNA is enzymatically fragmented to a size optimal for hybridization. The process is stopped, and the fragmentation efficiency can be checked, for example, by gel electrophoresis [44].
- Precipitation and Resuspension: The fragmented DNA is precipitated with isopropanol to concentrate and purify it. The resulting pellet is then resuspended in a hybridization buffer [44].
- Hybridization: The resuspended DNA is dispensed onto the Infinium BeadChip, which is then sealed and incubated in an oven (e.g., 48°C for 16–24 hours). During this time, the fragmented DNA anneals to the locus-specific probes on the beads [44] [45].
- Washing, Extension, and Staining (XStain): After hybridization, the BeadChip undergoes a series of automated washes to remove non-specifically bound and unhybridized DNA. This is followed by the critical single-base extension step, where the hybridized DNA acts as a template for the extension of the probe with a single fluorescently labeled dideoxynucleotide (ddNTP). The specific dye incorporated (corresponding to a C or T) indicates the methylation state. The extended products are then stained and coated for signal enhancement [6] [44] [45].
- Scanning: The processed BeadChip is imaged using an iScan System or similar scanner. The fluorescence intensities are captured and stored as data files (IDAT files) for subsequent analysis [44].
Data Analysis and Quality Control
After scanning, the raw data (IDAT files) must be processed to extract meaningful methylation values.
- Raw Data Extraction and Quality Control: Use software tools to assess data quality. DRAGEN Array Methylation QC software provides a high-throughput, quantitative report on 21 control metrics [9]. Alternatively, GenomeStudio Methylation Module or BeadArray Controls Reporter (BACR) can be used for basic QC, visualizing control probes and calculating detection p-values. Probes with a detection p-value > 0.01 are typically considered to have failed [9] [44].
- Normalization and Preprocessing: Due to the different signal intensities of Infinium I and II probes, normalization is essential. While GenomeStudio can perform initial normalization, it is not recommended for final analysis. Instead, use specialized R packages like SeSAMe or minfi, which implement advanced normalization techniques (e.g., NOOB - normal-exponential out-of-band) to correct for background and probe design bias [9] [46].
- Downstream Analysis: The normalized beta-values can be used for various analyses. For sperm epigenetics, this may include:
- Differential Methylation Analysis: Identifying CpG sites or regions (DMRs) that are significantly methylated differently between groups (e.g., fertile vs. infertile). Popular tools include
DMRcateandbumphunterin R [9] [46]. - Cell Type Deconvolution: Inferring cell type fractions, which in sperm research is crucial for confirming the absence of somatic cell contamination [9] [2].
- Epigenetic Age Prediction: Building or applying models to predict biological age from sperm methylation patterns, a growing area in forensic and reproductive science [7].
- Differential Methylation Analysis: Identifying CpG sites or regions (DMRs) that are significantly methylated differently between groups (e.g., fertile vs. infertile). Popular tools include
Table 2: Key Software Tools for Methylation Data Analysis
| Software/Package | Primary Function | Deployment | Key Feature |
|---|---|---|---|
| DRAGEN Array Methylation QC [9] | High-throughput Quality Control | Cloud (ICA) | 21 quantitative control metrics, detection p-values |
| GenomeStudio Methylation Module [9] | Visualization & Basic QC | Local (GUI) | Control plots, initial beta-value calculation |
| Partek Flow [9] | Downstream Multi-Omics Analysis | Cloud/Local (GUI) | Interactive statistics, differential methylation |
| SeSAMe [9] [46] | End-to-End Analysis & Normalization | R/Bioconductor | Improved normalization, QC, DMR calling |
| Minfi [9] [46] | Comprehensive Preprocessing & Analysis | R/Bioconductor | Data preprocessing, quality assessment |
| ChAMP [9] | EWAS Analysis Pipeline | R/Bioconductor | Integrated pre-processing, DMR, GSEA, visualization |
Troubleshooting and Technical Notes
- Reproducibility: The Infinium Methylation BeadChip demonstrates high technical robustness, with >98% reproducibility for technical replicates and high correlation with whole-genome bisulfite sequencing (WGBS) data [6] [46].
- Low DNA Input: While the official recommendation is 250 ng DNA input, studies have shown that the EPICv2 array can produce high-quality data with high correlation between replicates even with input levels below this recommendation [46].
- Cross-Hybridizing Probes: A known technical issue is that some probes can hybridize to multiple genomic locations. It is important to use updated manifest files and analysis pipelines (e.g., SeSAMe) that filter out these problematic probes to avoid inaccurate methylation quantification [46].
The analysis of DNA methylation in sperm epigenetics provides critical insights into male fertility, embryonic development, and transgenerational inheritance patterns. The Illumina Infinium Methylation BeadChip platform has emerged as a powerful tool for epigenome-wide association studies (EWAS) in this field, enabling the profiling of hundreds of thousands of CpG sites across the genome. The preprocessing of this data is a critical first step that significantly influences all downstream analyses and biological interpretations. Within this landscape, three prominent bioinformatic pipelines—minfi, ChAMP, and SeSAMe—have been developed to transform raw intensity data (.IDAT files) into reliable methylation values [48] [9]. Each pipeline offers distinct approaches to key preprocessing challenges including background correction, dye bias adjustment, normalization, and probe filtering. For sperm epigenetics research specifically, considerations such as the unique methylation patterns in germ cells, the impact of genetic variants on probe hybridization, and the need for accurate detection of imprinting control regions necessitate careful pipeline selection [7] [49]. This application note provides a detailed comparative analysis and experimental protocols for implementing these pipelines in the context of sperm epigenetics research, with a focus on practical implementation for researchers and drug development professionals.
Pipeline Architectures and Methodological Comparisons
Fundamental Preprocessing Concepts and Metrics
The Infinium BeadChip technology relies on a combination of probe designs (Type I and II) that require specialized processing to generate accurate, comparable methylation values [50]. The fundamental metrics include beta values (β = M/(M + U + 100)), representing the proportion of methylation at a CpG site ranging from 0 (completely unmethylated) to 1 (completely methylated), and M-values (log2(M/U)), which provide better statistical properties for differential methylation analysis [50]. Preprocessing aims to correct for technical artifacts including background noise, dye bias, batch effects, and probe-type differences while preserving biological signal [51] [52]. The success of these corrections is particularly important in sperm epigenetics, where subtle methylation changes at imprinting control regions can have significant functional consequences [49].
Comparative Analysis of Preprocessing Pipelines
The following table summarizes the key characteristics, strengths, and limitations of the three primary preprocessing pipelines:
Table 1: Comprehensive Comparison of DNA Methylation Preprocessing Pipelines
| Feature | minfi | ChAMP | SeSAMe |
|---|---|---|---|
| Primary Focus | General-purpose methylation analysis [50] | Comprehensive EWAS analysis [9] | Multi-species, artifact reduction [53] [52] |
| Core Normalization Methods | Subset-quantile within array normalization (SWAN), preprocessQuantile [51] | SWAN, BMIQ [9] | Noob (normal-exponential using out-of-band probes) [54] |
| Detection P-value Method | Combined background signals [52] | Combined background signals [9] | pOOBAH (P-value with Out-Of-Band Array Hybridization) [52] |
| Probe Filtering | SNP-associated, cross-reactive probes [50] | Automated filtering pipeline [9] | Genome-specific utility, SNP annotation [53] |
| Batch Effect Correction | ComBat integration [51] | ComBat integration [9] | Platform-aware preprocessing [55] |
| Sperm-Specific Considerations | Standard QC metrics | Standard QC metrics | SNP influence annotation for genetic variants [53] |
| Key Advantage | Established, widely validated | All-in-one EWAS solution | Reduced technical variation, improved cross-platform consistency [52] |
| Limitation | Less specialized for non-human genomes | Less optimized for multi-species | Steeper learning curve |
Experimental Protocols for Sperm Methylation Analysis
Sample Preparation and Quality Control for Sperm Epigenetics
For DNA methylation analysis of sperm samples, proper sample processing is essential to ensure data quality:
Sperm Isolation and Purification: Process semen samples using swim-up separation to isolate motile sperm. Centrifuge samples at 3,000 × g for 10 minutes, resuspend precipitate in PBS, and incubate at 37°C/5% CO₂ for 45-60 minutes to allow motile sperm migration. Confirm >99% purity via phase-contrast microscopy (20× magnification) to eliminate somatic cell contamination [49].
DNA Extraction and Quality Assessment: Extract genomic DNA using the QIAamp DNA Blood & Tissue Kit or similar. Quantify DNA purity using NanoDrop 260/280 and 260/230 ratios. Verify DNA integrity via agarose gel electrophoresis [49].
Bisulfite Conversion: Treat 500-1000 ng of DNA using the EZ DNA Methylation-Gold Kit or equivalent. Verify conversion efficiency through control probes on the array [48] [7].
Array Processing: Hybridize bisulfite-converted DNA (400 ng) to the Infinium HumanMethylation450K or EPIC BeadChip according to manufacturer's instructions [49].
Protocol 1: minfi Pipeline Implementation
Protocol 2: ChAMP Pipeline Implementation
Protocol 3: SeSAMe Pipeline Implementation
Benchmarking and Performance Evaluation
Comparative Performance in Sperm Epigenetics Studies
Multiple studies have evaluated the performance of preprocessing pipelines in various biological contexts, providing insights for sperm epigenetics research:
Table 2: Performance Metrics Across Preprocessing Pipelines
| Metric | minfi | ChAMP | SeSAMe |
|---|---|---|---|
| Technical Variation | Moderate [54] | Moderate [51] | Low [52] [54] |
| Cross-Platform Consistency | Moderate [54] | Moderate [54] | High [52] [55] |
| SNP Artifact Reduction | Basic filtering [50] | Basic filtering [9] | Advanced annotation [53] |
| Handling of Sample Degradation | Standard approach | Standard approach | Enhanced detection calling [52] |
| Computational Efficiency | Moderate | Moderate | High [52] |
In a direct comparison evaluating data harmonization between 450K and EPIC platforms, SeSAMe normalization demonstrated superior performance in technical replicate concordance, with tighter distribution of absolute differences in beta values compared to SWAN normalization (commonly used in minfi) [54]. The pOOBAH detection method in SeSAMe specifically addresses hybridization failures due to germline deletions or hyperpolymorphism, which is particularly valuable in sperm epigenetics where genetic variants can influence methylation readings [53] [52].
Quality Control Metrics for Sperm-Specific Analyses
Implementation of rigorous QC measures is essential for robust sperm methylation studies:
Detection P-values: Filter probes with detection p > 0.01 in >5% of samples across all pipelines [52].
Bisulfite Conversion Controls: Verify conversion efficiency >99% through built-in control probes.
Sex Chromosome Profiling: Confirm sample sex consistency through chromosome X/Y methylation patterns—particularly important for verifying sperm sample purity [52].
Technical Replicates: Include cross-platform replicates to evaluate data harmonization when combining datasets [54].
Somatic Cell Contamination Check: Assess for abnormal methylation patterns at imprinted loci that may indicate somatic cell contamination in sperm samples [49].
Research Reagent Solutions for Sperm Methylation Analysis
Table 3: Essential Research Reagents for Sperm Methylation Studies
| Reagent/Kit | Function | Application Note |
|---|---|---|
| QIAamp DNA Blood & Tissue Kit | Genomic DNA extraction | High-quality DNA extraction from sperm samples [49] |
| EZ DNA Methylation-Gold Kit | Bisulfite conversion | Efficient cytosine-to-uracil conversion [7] [49] |
| Infinium HD Assay Methylation Kit | BeadChip processing | Library preparation for array hybridization |
| NucleoMag DNA Blood Kit | High-throughput DNA extraction | Suitable for large-scale epidemiological studies |
| Illumina HumanMethylationEPIC v2 | Methylation profiling | Coverage of >935,000 CpG sites including enhancer regions [55] |
Workflow Visualization and Decision Pathways
Preprocessing Pipeline Selection Algorithm
Integrated Sperm Methylation Analysis Workflow
Based on comprehensive evaluation of the three preprocessing pipelines, we recommend the following for sperm epigenetics research:
For studies prioritizing artifact reduction and multi-platform consistency: Implement SeSAMe pipeline with pOOBAH detection calling, which specifically addresses hybridization failures and demonstrates superior technical performance in comparative studies [52] [54].
For all-in-one EWAS analysis with standardized workflows: Utilize ChAMP pipeline, which provides integrated functionality for the entire analysis workflow from preprocessing to DMP/DMR detection [9].
For established methodologies with extensive community usage: Apply minfi pipeline with preprocessQuantile normalization, particularly when comparing with existing published datasets [50].
For sperm-specific considerations: Implement additional quality checks for somatic cell contamination and verify imprinting region methylation patterns, regardless of pipeline selection [49].
The emerging EPICv2 array presents new opportunities for enhanced coverage of regulatory elements in sperm epigenetics studies. When combining data across different array versions, apply platform-specific normalization followed by meta-analysis or explicit version adjustment in statistical models to mitigate technical variability [55]. As sperm epigenetics continues to advance in understanding heritable epigenetic patterns and their implications for offspring health, appropriate preprocessing methodologies will remain fundamental to generating biologically meaningful results.
The Infinium Methylation BeadChip has established itself as a cornerstone technology for epigenome-wide association studies, offering a cost-effective, quantitative, and user-friendly platform for profiling DNA methylation [4]. Within the specialized field of sperm epigenetics, this technology enables researchers to decipher the complex epigenetic signatures associated with male fertility, environmental exposures, and transgenerational inheritance. This application note details advanced methodologies for two cutting-edge applications: the construction and implementation of sperm epigenetic clocks to measure biological age, and sophisticated computational deconvolution approaches to address cellular heterogeneity in semen samples. These protocols provide a critical framework for ensuring data accuracy and biological relevance in male reproductive epigenetic studies, supporting both basic research and clinical applications in reproductive medicine.
Sperm Epigenetic Clocks: Measuring Biological Age
Concept and Clinical Utility
The sperm epigenetic clock is a biomarker that captures the biological age of sperm, which may differ from chronological age and provide superior predictive value for reproductive outcomes. Chronological age serves as a proxy for reproductive capacity but fails to encapsulate cumulative genetic and environmental factors that constitute the 'true' biological age of cells [56]. Research demonstrates that sperm epigenetic aging clocks act as a novel biomarker to predict a couple's time to pregnancy. Studies have found a 17% lower cumulative probability of pregnancy after 12 months for couples with male partners in older sperm epigenetic aging categories compared to those with younger epigenetic ages [56]. Furthermore, higher sperm epigenetic aging is associated not only with longer time to pregnancy but also with shorter gestation periods in couples that achieve pregnancy [56].
Development and Validation
The development of a sperm epigenetic clock involves sophisticated computational modeling of DNA methylation data derived from Infinium Methylation BeadChips. A recent mouse study established a sperm epigenetic clock model to evaluate effects of interventions on DNA methylome aging, identifying that environmental stressors like heat stress and cadmium exposure can accelerate epigenetic aging of sperm via mTOR/Blood-Testis Barrier mechanisms [57]. In human applications, these clocks are built using machine learning algorithms trained on methylation data from donors of known chronological age, with validation in independent cohorts.
The following table summarizes key quantitative findings from recent sperm epigenetic clock studies:
Table 1: Quantitative Findings from Sperm Epigenetic Clock Studies
| Study Model | Key Measurement | Value/Outcome | Clinical/Biological Significance |
|---|---|---|---|
| Human Cohort [56] | Pregnancy Probability Reduction | 17% lower | Associated with older sperm epigenetic age |
| Mouse Model [57] | Testis Weight Reduction (34.5°C heat stress) | ~20% decrease (100.3mg to 80.2mg) | Indicator of stressor impact on testicular function |
| Mouse Model [57] | Testis Weight Reduction (Cadmium) | ~26% decrease (100.3mg to 74.1mg) | Indicator of toxicant impact on testicular function |
Protocol: Developing a Sperm Epigenetic Clock
Materials:
- Infinium MethylationEPIC BeadChip Kit (Illumina)
- DNeasy Blood/Tissue Kit (Qiagen) or equivalent DNA extraction system
- EZ-96 DNA Methylation Kit (Zymo Research)
- Laboratory equipment: TissueLyser II, Nanodrop spectrophotometer, iSCAN reader
Procedure:
- Sample Collection and Preparation: Collect semen samples from a cohort of male donors spanning a wide age range (e.g., 21-69 years) with documented fertility status. Obtain informed consent and ethical approval.
- Sperm Isolation and DNA Extraction:
- Isolate motile sperm using the swim-up method to minimize somatic cell contamination [58]. Layer the semen sample under wash medium (Earle's Balanced Salt Solution with HEPES and human albumin) and incubate at 37°C at a 45° angle for 2 hours. Harvest sperm from the supernatant.
- Extract DNA using the DNeasy Blood/Tissue Kit with modifications for sperm: add steel beads and homogenize using a TissueLyser II, followed by overnight incubation with proteinase K and RNAse A treatment [58].
- DNA Methylation Profiling:
- Bioinformatic Analysis and Clock Building:
- Process raw IDAT files using the SeSAMe pipeline in R, which includes quality control, dye bias correction, background subtraction, and detection p-value filtering [58].
- Use the
minfiorewastoolspackages to calculate beta values (β) representing methylation levels (0-1 scale). - Divide the cohort into training (e.g., 70%) and validation (e.g., 30%) sets.
- In the training set, apply an elastic net regression model (via the
glmnetR package) to identify a panel of CpG sites whose methylation levels collectively predict chronological age. This penalized regression selects the most predictive CpGs while avoiding overfitting. - Validate the resulting model in the hold-out validation set by calculating the correlation (Pearson's r) between predicted epigenetic age and chronological age, and the median absolute error.
- Application: The finalized model can be applied to new samples to obtain their sperm epigenetic age (SEA). A positive age acceleration residual (difference between SEA and chronological age) indicates a biologically older sperm epigenome.
Figure 1: Workflow for developing a sperm epigenetic clock, from sample collection to validated model.
Addressing Cellular Heterogeneity: Deconvolution in Sperm Samples
The Challenge of Somatic Cell Contamination
Semen samples represent a complex cellular mixture containing the sperm of interest but also potentially significant numbers of somatic cells, such as leukocytes and epithelial cells. This contamination poses a major challenge for sperm-specific epigenetic analysis because somatic cells have distinctly different DNA methylation profiles [20]. In healthy normozoospermic men, somatic cells may be present at concentrations up to 1×10^6 cells/ml of semen, with this number increasing substantially in oligozoospermic individuals [20]. Critically, even low-level contamination (e.g., 5%) can significantly bias DNA methylation measurements, as hypermethylation at specific loci might be misinterpreted as a sperm-specific epigenetic alteration when it actually originates from contaminating somatic cells [20].
Deconvolution Strategies
Two primary strategies exist to manage this contamination:
- Wet-Lab Purification: Physical separation of sperm from somatic cells prior to DNA extraction.
- Computational Deconvolution: In silico separation of methylation signals after data generation.
A robust research plan incorporates both approaches. The most effective method involves initial purification steps followed by computational verification to eliminate any residual confounding influence.
Protocol: A Comprehensive Plan to Eliminate Somatic DNA Contamination
Materials:
- Somatic Cell Lysis Buffer (SCLB): 0.1% SDS, 0.5% Triton X-100 in ddH₂O
- Phosphate Buffered Saline (PBS), pH 7.4
- Refrigerated centrifuge
- Inverted microscope (e.g., Nikon Eclipse Ti-S with 20X objective)
Procedure:
Part A: Wet-Lab Somatic Cell Removal
- Initial Wash and Inspection:
- Wash fresh semen samples twice with 1X PBS by centrifugation at 200 g for 15 minutes at 4°C.
- Resuspend the pellet and inspect an aliquot under a microscope to identify the level of somatic cell contamination and perform a sperm count [20].
- Somatic Cell Lysis:
- Incubate the washed sample with freshly prepared Somatic Cell Lysis Buffer (SCLB) for 30 minutes at 4°C [20].
- Centrifuge to pellet the cells.
- Post-Lysis Inspection:
- Re-examine the sample under a microscope to confirm the absence or significant reduction of somatic cells and repeat the sperm count.
- If somatic cells are still detected, repeat the SCLB treatment.
- Final Sperm Pellet:
- If no somatic cells are detected, pellet the pure sperm population by centrifugation and perform a final wash with PBS before DNA extraction [20].
Part B: Computational Quality Control and Deconvolution
- Utilize Established Somatic Methylation Markers:
- Following DNA methylation profiling with the BeadChip, screen your data against a predefined panel of 9,564 CpG sites that have been identified as highly specific markers for somatic DNA contamination. These sites exhibit >80% methylation in blood cells but <20% methylation in pure sperm and are not linked to infertility [20].
- Calculate Contamination Estimate:
- The aggregate methylation signal across these marker CpGs provides an estimate of the level of residual somatic contamination in the sample.
- Apply Analysis Threshold:
- Implement a 15% methylation cut-off during differential methylation analysis. This conservative threshold, informed by modeling various contamination scenarios, helps prevent false positive calls of hypermethylation that are actually driven by somatic cell signals [20].
Figure 2: A dual-phase workflow combining physical somatic cell removal with computational quality control to ensure pure sperm methylation data.
Advanced Computational Deconvolution Methods
For researchers analyzing complex tissues like the testis, advanced computational deconvolution methods can be invaluable. Reference-free deconvolution methods are particularly powerful as they do not require purified cell type profiles as a reference, which are often unavailable.
SURF (Self-sUpervised Deep Learning Reference-Free method) is a state-of-the-art tool designed for spot-level spatial transcriptomic data that can be adapted for methylation data analysis. It employs an autoencoder architecture to model nonlinear gene interactions and uses contrastive learning to incorporate relationships between spots (or samples) [59]. Spatially adjacent spots with high gene expression similarities are pulled closer in the model, leading to similar cell-type composition predictions, while spots with significant disparities are pushed apart [59]. This approach has demonstrated superior performance in accurately recovering cell-type compositions compared to other reference-free methods, especially when appropriate single-cell references are lacking [59].
Table 2: Key Reagent Solutions for Sperm Epigenetics Studies
| Research Reagent / Tool | Specific Function | Application Context |
|---|---|---|
| Infinium MethylationEPIC BeadChip v2 | Genome-wide DNA methylation profiling at ~935,000 CpG sites. | Core platform for generating sperm methylome data for clock building and differential methylation analysis [4]. |
| Somatic Cell Lysis Buffer (SCLB) | Selectively lyses contaminating somatic cells (e.g., leukocytes) while preserving sperm integrity. | Critical wet-lab step for purifying sperm cells from raw semen prior to DNA extraction [20]. |
| Swim-Up Media (Earle's Balanced Salt Solution + HEPES + Human Albumin) | Isolates a highly motile, viable fraction of sperm, further reducing somatic cell carryover. | Sperm purification protocol to enrich for functional sperm and improve sample purity [58]. |
| SeSAMe (Preprocessing Pipeline) | Processes raw IDAT files: performs quality control, dye bias correction, and background subtraction. | Essential bioinformatic tool for standardizing and cleaning methylation array data before analysis [58]. |
| SURF Algorithm | Reference-free deconvolution using self-supervised deep learning. | Advanced computational tool for inferring cell-type proportions in mixed samples, useful for complex testicular tissue [59]. |
| Somatic CpG Marker Panel (9,564 CpGs) | A predefined set of genomic loci hypermethylated in blood/soma but hypomethylated in sperm. | Computational quality control step to estimate and flag residual somatic contamination in processed sperm samples [20]. |
The integration of Infinium Methylation BeadChip technology with robust experimental and computational protocols for epigenetic clocking and cell deconvolution significantly advances the field of sperm epigenetics. The methods detailed herein—ranging from meticulous wet-lab purification to sophisticated computational checks and the application of novel algorithms like SURF—provide researchers with a comprehensive toolkit to generate high-quality, biologically meaningful data. These approaches are crucial for accurately linking sperm DNA methylation patterns to male fertility, offspring health, and the impacts of environmental exposures, ultimately driving discovery in reproductive biology and medicine.
Maximizing Data Fidelity: Tackling Technical Noise and Probe Reliability
The Infinium MethylationEPIC BeadChip is a powerful tool for epigenome-wide association studies (EWAS) in sperm epigenetics research, enabling insights into male fertility, environmental exposures, and transgenerational inheritance [2] [60]. However, technical challenges during the experimental workflow can compromise data quality and reliability. This application note addresses three common laboratory challenges—precipitate formation, bubble formation, and BeadChip drying issues—within the context of sperm epigenetic profiling. We provide detailed protocols and quantitative data to help researchers mitigate these issues, ensuring robust and reproducible results for drug development and clinical research.
Troubleshooting Common Challenges
Precipitate in Hybridization Solution
Observation: A small to large amount of precipitate is visible in the hybridization solution.
Table 1: Troubleshooting Precipitate Formation
| Symptom | Probable Cause | Resolution / Comment |
|---|---|---|
| Small amount of precipitate | Normal occurrence in hybridization solution | Does not affect data quality; continue with the experiment [61] [62]. |
| Large, unresuspended precipitate | Excessive evaporation after heat denaturing due to improper sealing | Use a foil heat sealer for all temperatures ≥ 45°C; ensure the sealer is properly seated to prevent evaporation. If precipitate cannot be resuspended, the sample may be compromised [61]. |
Bubble Formation and Air Pockets
Observation: Air bubbles prevent proper pellet dissolution or create uncoated areas on BeadChips.
Table 2: Troubleshooting Bubble and Air Pocket Formation
| Symptom | Probable Cause | Resolution / Comment |
|---|---|---|
| Blue pellet does not dissolve after vortexing | Air bubble trapped at the bottom of the well | Pulse centrifuge the plate to 280 × g to remove the bubble, then revortex at 1800 rpm for 1 minute [61]. |
| Solution foams excessively during dispensing | Pipetting was too vigorous | Pipette gently to avoid creating bubbles. Centrifuge the plate to 280 × g to remove existing bubbles [61]. |
| Uncoated areas on BeadChip after XC4 coating | Bubble formed during coating, preventing solution contact | Briefly place the staining rack back into the XC4 wash dish. Gently move BeadChips back and forth while moving up and down to break the bubble [61] [62]. |
BeadChip Drying Issues
Observation: BeadChips remain wet after vacuum desiccation or show unusual reagent flow.
Table 3: Troubleshooting BeadChip Drying and Flow Issues
| Symptom | Probable Cause | Resolution / Comment |
|---|---|---|
| BeadChips still wet after 55 minutes in vacuum desiccator | Lab temperature/humidity, old XC4, or old ethanol | Extend drying time. Replace XC4 (reusable up to six times in two weeks). Replace ethanol with a fresh bottle, as old ethanol may have absorbed atmospheric water [61] [62]. |
| Liquid in Flow-Through Chamber drops below reservoir | Dirty glass backplates, incorrect spacer, or insecure assembly | Thoroughly clean glass backplates before and after each use. Ensure the correct spacer is used and that the Flow-Through Chamber is securely assembled with metal clamps [62]. |
| Unusual reagent flow patterns | Residue build-up on glass backplates | Clean glass backplates thoroughly before and after use to remove protein, enzyme, or antibody residue [62]. |
Sperm Epigenetics-Specific Protocols
Comprehensive Protocol for Mitigating Somatic Cell Contamination
A primary concern in sperm epigenetics is the confounding effect of somatic DNA contamination on methylation data [2]. The following integrated protocol is essential for generating meaningful data.
Title: Sperm QC and contamination mitigation workflow
1. Initial Quality Check: Microscopic Examination
- Visually inspect the crude semen sample under a microscope to assess the initial presence of somatic cells [2].
2. Somatic Cell Lysis
- Treat the semen sample with a Somatic Cell Lysis Buffer (SCLB) to selectively lyse contaminating white blood cells and other somatic cells, preserving the sperm cells [2].
3. Sperm DNA Isolation
- Isolate sperm DNA using a protocol designed for protamine-bound DNA. A recommended method involves:
- Lysis Buffer: Guanidine thiocyanate and 50 mM tris(2-carboxyethyl) phosphine (TCEP). TCEP is a stable reducing agent that breaks protamine disulfide bonds [60].
- Homogenization: Use 0.2 mm steel beads for efficient lysis [60].
- Purification: Use silica-based spin columns. This rapid method works at room temperature, avoids lengthy proteinase K digestions, and yields high-quality DNA [60].
4. Infinium Assay Processing
- Process the purified sperm DNA using the standard Infinium MethylationEPIC BeadChip protocol, remaining vigilant for the common challenges outlined in Section 2 [47] [60].
5. Post-Hybridization Quality Control
- Analyze Somatic Contamination Markers: Compare your data to a reference set of 9,564 CpG sites that are highly methylated in blood compared to sperm. These serve as biomarkers for somatic contamination [2].
- Apply a Contamination Cut-off: Exclude samples where the methylation pattern suggests a somatic DNA contamination level of >15% to draw error-free scientific conclusions [2].
Low-Input DNA Considerations for Rare Sperm Samples
For samples with limited DNA, such as those from oligospermic men, the standard 250 ng input requirement for the Infinium assay can be prohibitive. Recent methodological advances offer potential solutions.
- DNA Preamplification: Incorporating a whole-genome amplification step prior to the bisulfite conversion and Infinium assay can significantly enhance detection rates, enabling analysis of samples with DNA inputs as low as five cells [63].
- Enzymatic Conversion: Using enzymatic conversion instead of sodium bisulfite can better preserve DNA integrity, improving data quality from low-input samples [63].
- Computational Correction: New signal detection frameworks have been developed to model background noise in low-input data, maximizing the retention of true biological signals that might otherwise be masked by conservative probe-filtering thresholds [63].
The Scientist's Toolkit
Table 4: Essential Research Reagent Solutions for Sperm Epigenetics
| Item | Function / Application | Specifications / Notes |
|---|---|---|
| Somatic Cell Lysis Buffer (SCLB) | Selective lysis of non-sperm cells in semen samples to minimize somatic DNA contamination for accurate sperm methylome analysis [2]. | Critical for pre-processing semen samples prior to DNA extraction. |
| Tris(2-carboxyethyl)phosphine (TCEP) | A stable, room-temperature reducing agent used in sperm DNA lysis buffers to break protamine disulfide bonds and efficiently release DNA [60]. | Preferred over volatile agents like DTT or BME. |
| Infinium HD FFPE DNA Restoration Kit | Restores DNA that has been fragmented or damaged, which can be useful for low-quality samples or when adapting low-input protocols [63]. | Not part of the standard protocol but valuable for challenging samples. |
| Infinium MethylationEPIC BeadChip Kit | Genome-wide profiling of DNA methylation at over 850,000 CpG sites. The primary tool for sperm epigenome-wide association studies (EWAS) [47] [60]. | Requires iScan System for scanning. |
| Foil Heat Sealer | Ensures a secure seal on assay plates during high-temperature incubation steps (≥45°C), preventing evaporation that leads to precipitate formation [61] [62]. | Essential for preventing sample loss during heat denaturation. |
| XC4 Coating Solution | A solution used in the XStain process to prepare BeadChips for imaging. Must be fresh to ensure proper drying and performance [61]. | Reusable up to six times within a two-week period [62]. |
In sperm epigenetics research, the Infinium Methylation BeadChip platform serves as a vital tool for probing DNA methylation landscapes. A significant technical challenge in this domain is the presence of unreliable probes, with low signal intensity being a primary contributor. These problematic probes can introduce substantial variability and bias, potentially obscuring true biological signals and compromising the validity of epigenetic findings in sperm studies [64]. This application note delineates the impact of low signal intensity on data reliability and provides a detailed, actionable protocol for the identification and filtration of unreliable probes, thereby ensuring robust and reproducible results in methylation analyses of sperm-derived DNA.
Quantitative Impact of Low Signal Intensity
Low signal intensity is a critical determinant of probe performance on Infinium BeadChips. Probes with low mean intensity (MI) exhibit significantly higher variability in methylation β values between technical replicates [64]. This relationship is quantifiable, and the following table summarizes key metrics and thresholds related to probe unreliability.
Table 1: Key Quantitative Metrics for Identifying Unreliable Probes Based on Signal Intensity
| Metric | Description | Impact on Reliability | Recommended Threshold |
|---|---|---|---|
| Mean Intensity (MI) | The average signal intensity from the methylated and unmethylated channels for a probe [64]. | Probes with low MI show higher β value variability between replicates [64]. | Dynamic, dataset-specific thresholding is recommended [64]. |
| Unreliability Score | A score estimated by simulating the influence of technical noise on β values using negative control probe backgrounds [64]. | Higher scores indicate greater susceptibility to technical noise; correlates negatively with MI [64]. | Use dynamic thresholds derived from probe-level simulation [64]. |
| DNA Input Quantity | Total DNA mass used for the BeadChip assay [65]. | Inputs as low as 40ng are feasible but increase noise and reduce power; below this, quality deteriorates markedly [65]. | 250ng (manufacturer's recommendation); 40ng is a functional lower limit with quality checks [65]. |
| Number of C-bases | The quantity of cytosine bases in the probe sequence [64]. | A higher number is associated with lower MI, providing a sequence-level predictor of potential unreliability [64]. | Consider as a factor during probe evaluation and filtering. |
Experimental Protocol for Assessing and Filtering Unreliable Probes
This protocol provides a step-by-step methodology for evaluating probe reliability based on signal intensity and for implementing an effective filtering strategy.
Sample Preparation and Data Generation
- DNA Extraction and Bisulfite Conversion: Extract DNA from sperm samples using a standardized phenol-chloroform or column-based kit. Assess DNA quality and quantity via spectrophotometry (e.g., NanoDrop) or fluorometry (e.g., Qubit). Perform bisulfite conversion on a minimum of 250 ng of DNA using a commercial kit (e.g., Zymo Research EZ DNA Methylation-Lightning Kit) to ensure optimal results, though inputs down to 40 ng can be used with an expected increase in noise [65].
- Methylation Array Processing: Process the bisulfite-converted DNA on the chosen Infinium Methylation BeadChip (e.g., EPIC v1.0, EPIC v2.0) strictly according to the manufacturer's protocol. Scan the arrays using the iScan System.
- Data Import and Raw Data Extraction: Import raw data files (
.idatformats) into an analysis environment like R. Use packages such asminfiorSeSAMeto read the intensity data and calculate raw methylation β values using the formula:β = M / (M + U + 100), where M and U represent methylated and unmethylated signal intensities, respectively [64].
Probe Reliability Assessment and Filtering Workflow
Calculation of Mean Intensity (MI) and Unreliability Scores:
- For each probe, calculate the Mean Intensity (MI). The specific computational method may be implemented in specialized R packages [64].
- Calculate an unreliability score for each probe by simulating the effect of technical noise on the β value. This simulation leverages the background intensity distribution of negative control probes on the array to model how technical variation propagates into methylation measurement uncertainty [64].
Establishment of Dynamic Thresholds:
- Avoid using fixed, universal thresholds for MI and unreliability scores. Instead, determine dynamic, dataset-specific thresholds based on the distribution of these metrics across all probes in your experiment [64].
- This approach accounts for technical variations specific to your sample processing and batch conditions.
Filtering of Unreliable Probes:
- Remove probes with MI and unreliability scores that fall beyond the established dynamic thresholds.
- Integrate this intensity-based filtering with standard pre-processing steps, including the removal of probes containing single nucleotide polymorphisms (SNPs), those with demonstrated cross-reactivity, and those located on sex chromosomes if not relevant to the study design [64] [4].
Data Normalization and Downstream Analysis:
- Perform intra-array normalization (e.g., using
minfi::preprocessFunnormorChAMP::champ.norm) to correct for technical biases between different probe types (Infinium I and II) [64]. - Proceed with differential methylation analysis and other downstream applications using the filtered and normalized dataset.
- Perform intra-array normalization (e.g., using
The following diagram illustrates the core logical workflow for this protocol:
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for Reliable Methylation Analysis
| Item | Function / Rationale |
|---|---|
| Infinium MethylationEPIC BeadChip | Microarray platform for genome-wide methylation profiling at over 850,000 (v1.0) or ~930,000 (v2.0) CpG sites. The newer v2.0 offers improved coverage but retains some probes with poor reliability scores [64] [25]. |
| PAXgene Blood DNA Tubes / Sperm Storage Buffer | For standardized and stable collection and preservation of biological samples, preventing degradation of DNA. |
| DNA Extraction Kit (e.g., Machery Nagel NucleoMag) | For high-quality DNA extraction from sperm cells, a critical step for reliable downstream bisulfite conversion and array hybridization [64]. |
| Bisulfite Conversion Kit (e.g., Zymo Research EZ-96 DNA Methylation-Lightning) | Chemically converts unmethylated cytosines to uracils, enabling methylation status discrimination. Efficiency is paramount for data quality [64] [37]. |
| R Package: 'minfi' or 'ChAMP' | Bioinformatics tools for primary data pre-processing, including background correction, normalization, and initial quality control [64] [66]. |
| Custom R Package for Unreliability Scores | A specialized tool for calculating MI and unreliability scores, facilitating data-driven probe filtering as described in the protocol [64]. |
| SeSAMe Package | An alternative pre-processing pipeline that includes methods for background subtraction and dye bias correction, with recent versions supporting multi-species and enhanced probe utility analysis [53] [6]. |
Addressing Probe Cross-Reactivity and Genetic Variation (SNP) Interference
The Infinium Methylation BeadChip represents a widely adopted technology for genome-wide DNA methylation analysis in sperm epigenetics research, offering a cost-effective and quantitative approach for characterizing epigenetic marks [4]. However, two significant technical challenges—probe cross-reactivity and genetic variation interference—can compromise data integrity if not properly addressed. Cross-reactivity occurs when probes hybridize to multiple genomic locations, while single nucleotide polymorphisms (SNPs) at or near target CpG sites can interfere with probe hybridization and methylation quantification. These issues are particularly relevant in sperm epigenetics studies, where accurate methylation measurement is crucial for understanding paternal age effects, infertility, and transgenerational epigenetic inheritance [26] [67] [29]. This application note provides comprehensive guidance for identifying and mitigating these technical artifacts to ensure data quality in studies utilizing the Infinium MethylationEPIC v2 BeadChip (EPICv2) and its predecessors.
Understanding Probe Design and Technical Artifacts
Infinium BeadChip Probe Chemistry and Evolution
The Infinium methylation technology utilizes bisulfite-converted DNA, which converts unmethylated cytosines to uracils, creating a C/T variant that can be interrogated using microarray technology [68]. The platform employs two distinct probe chemistries: Infinium-I uses two separate bead types (methylated and unmethylated alleles), while Infinium-II utilizes a single bead type with color discrimination to distinguish methylation states [4]. The evolution from HM450 to EPICv1 and subsequently to EPICv2 has brought significant improvements in probe design, with EPICv2 featuring 937,690 probes and demonstrating better mapping efficiency to the GRCh38 reference genome compared to its predecessors [4].
Recent evaluations of EPICv2 have revealed substantial improvements in addressing technical artifacts. Specifically, EPICv2 contains fewer probes with poor mapping characteristics and reduced susceptibility to direct influence by ancestry-specific genetic variation [4]. Of the probes deleted in EPICv2, 72.9% were found to have issues with cross-reactivity or direct influence from sequence polymorphism, compared to only 0.1% of retained probes [4]. These design enhancements result in more accurate methylation assessment across diverse human populations, though careful quality control remains essential.
Mechanisms of Cross-Reactivity and SNP Interference
Cross-reactivity represents a major technical artifact where probes hybridize to multiple genomic locations, measuring a mixture of specific and aspecific signals [68]. This phenomenon can lead to spurious associations in epigenome-wide association studies (EWAS), particularly when cross-hybridization targets regions with structural variation or repeat expansions associated with the phenotype of interest [68].
SNP interference occurs when genetic variations at or near the target CpG site affect probe hybridization efficiency. SNPs can directly prevent probe binding through sequence mismatch or create additional CpG sites that confound methylation measurement [4] [55]. The impact of SNP interference varies across human populations, with African ancestry groups typically showing greater effects due to higher genetic diversity [4].
Table 1: Characteristics of Problematic Probes in Infinium Methylation BeadChips
| Probe Issue Type | Mechanism | Impact on Data Quality | Frequency in EPICv1 | Improvement in EPICv2 |
|---|---|---|---|---|
| Cross-reactive probes | Hybridization to multiple genomic locations | Inflated background signal, spurious associations | ~6-11% of probes | Significant reduction through improved design |
| SNP-affected probes | Genetic variation at target site | Altered hybridization efficiency, false methylation calls | Varies by ancestry | Fewer probes subject to ancestry-specific variation |
| Poorly mapping probes | Non-unique alignment in reference genome | Inaccurate methylation quantification | Substantial number | Improved mapping to GRCh38 |
| Strand switch probes | Incorrect strand specification | Systematic measurement bias | Present in EPICv1 | 22 probes with corrected strand choice |
Experimental Protocols for Quality Control
Comprehensive Probe Filtering Workflow
A rigorous probe filtering protocol is essential before any analytical application of methylation data. The following workflow should be implemented using R/Bioconductor packages:
Step 1: Initial Quality Assessment
- Calculate detection p-values for all probes across all samples
- Remove probes with detection p-value > 1×10⁻¹⁶ in >5% of samples [68]
- Eliminate probes measured by <3 beads in >5% of samples [68]
- Remove samples with >5% of probes failing detection thresholds [68]
Step 2: Cross-reactivity Filtering
- Annotate probes using updated cross-reactivity databases
- Remove probes with multiple alignments to the reference genome
- Flag probes with near matches (≤30 bp) to off-target regions [68]
- Pay special attention to probes targeting regions with structural variation
Step 3: SNP Filtering
- Annotate probes containing SNPs in the probe binding site
- Utilize population-specific SNP frequency data (e.g., 1000 Genomes Project)
- Remove probes with MAF >0.01 in the study population
- Consider using genetic matching between methylation data and WGS-derived SNPs [68]
Step 4: Additional Filtering Steps
- Remove non-CpG probes ("ch" probes) unless specifically needed
- Eliminate probes with known manufacturing issues
- Filter sex chromosome probes if analyzing autosomal methylation only
- Apply population-specific filters for multi-ethnic studies
Table 2: Recommended Quality Control Thresholds for Methylation Data
| QC Metric | Threshold | Software Implementation | Rationale |
|---|---|---|---|
| Sample detection rate | <5% failed probes | minfi, sesame | Identifies poor-quality samples |
| Probe detection rate | <5% failed samples | minfi, sesame | Identifies poorly performing probes |
| Cross-reactive probes | Complete removal | Predefined annotation files | Eliminates multi-mapping probes |
| SNP-containing probes | MAF >0.01 | minfi, FDb.InfiniumMethylation.hg19 | Reduces genetic confounding |
| Bead count | <3 beads | minfi, sesame | Ensures measurement precision |
| Sex mismatch | Discordance check | minfi getSex function | Identifies sample mix-ups |
Data Preprocessing and Normalization
Following probe filtering, appropriate normalization is critical:
- Perform background correction using out-of-band probes for Infinium-I probes [4]
- Apply dye bias correction using normalization controls
- Implement between-array normalization (e.g., quantile normalization)
- For EPICv2 data, utilize the SeSAMe package for optimal processing [69]
- Consider the ELBAR algorithm for suboptimal DNA input samples instead of pOOBAH [69]
Visualization of Quality Control Workflow
Quality Control Workflow for Methylation Data
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Computational Tools for Quality Control
| Resource | Type | Function | Application Notes |
|---|---|---|---|
| Zymo Bisulfite Conversion Kit | Wet-bench reagent | Converts unmethylated cytosines to uracils | Essential for methylation array preprocessing |
| Illumina EPICv2 BeadChip | Microarray | Genome-wide methylation profiling | Improved probe design reduces artifacts |
| minfi R/Bioconductor package | Software | Comprehensive methylation data analysis | Standard for QC and preprocessing |
| SeSAMe R/Bioconductor package | Software | Specific processing for EPICv2 data | Implements improved normalization methods |
| sesame R/Bioconductor package | Software | Quality control and analysis | Compatible with custom arrays [70] |
| OMICsPrint package | Software | Sample identity verification | Checks concordance with genotype data [68] |
| InfiniumAnnotation files | Annotation resource | Probe mapping and annotation | Essential for cross-reactivity and SNP filtering |
| ELBAR algorithm | Computational method | Detection calling for low-input DNA | Alternative to pOOBAH for degraded samples [69] |
Special Considerations for Sperm Epigenetics Research
Sperm DNA methylation exhibits unique characteristics that necessitate specialized analytical approaches. The sperm epigenome is fundamentally different from somatic cells, with distinct methylation patterns established during germ cell development [67]. Several factors require particular attention in sperm methylation studies:
Paternal Age Effects: Advanced paternal age is associated with systematic methylation changes in sperm, with approximately 74% of age-related differentially methylated regions (ageDMRs) showing hypomethylation and 26% hypermethylation [29]. These changes predominantly affect genes involved in development and nervous system function [26] [29].
Imprinted Genes: Sperm methylation analysis requires careful handling of imprinted genes, which maintain parent-of-origin specific methylation patterns. The Human Imprintome array represents a specialized tool for comprehensive assessment of imprint control regions (ICRs) [70].
Sample Quality Considerations: Sperm samples may yield limited DNA quantity, requiring optimized protocols. EPICv2 supports DNA input down to 1ng, though 250ng is recommended [4]. For fragmented DNA samples (e.g., from FFPE tissue or cfDNA), performance decreases significantly with average fragment sizes below 165bp [69].
Addressing probe cross-reactivity and SNP interference is essential for generating high-quality methylation data from sperm samples. The implementation of rigorous quality control protocols, utilizing updated annotation resources, and understanding platform-specific limitations will significantly enhance data reliability. Researchers should:
- Always apply comprehensive probe filtering before analysis
- Utilize version-specific annotations and processing methods
- Consider population genetic structure when applying SNP filters
- Implement specialized approaches for sperm-specific epigenetic features
- Maintain consistency in preprocessing across study samples
Following these guidelines will improve the accuracy of methylation quantification in sperm epigenetics research, enabling more robust investigations into paternal epigenetic contributions to development and disease.
Strategies for Data Normalization and Background Correction
The application of Infinium Methylation BeadChip technology to sperm epigenetics research presents unique bioinformatic challenges, necessitating robust strategies for data normalization and background correction. DNA methylation analysis in sperm cells is complicated by their distinct epigenetic landscape compared to somatic tissues, including globally hypomethylated regions and different age-related methylation patterns [37]. Effective normalization is critical to account for technical variance arising from multiple sources, including bisulfite conversion efficiency, sample processing dates, individual array positions on slides, and the fundamental differences between Infinium I and Infinium II probe chemistries [71]. These technical artifacts can artificially inflate within-group variances, reduce experimental power, and potentially create false positive results in epigenome-wide association studies (EWAS) if not properly addressed [71]. This protocol outlines comprehensive strategies tailored specifically for sperm methylation data, enabling accurate detection of biological signals amidst technical noise for research and potential clinical applications in male fertility and reproductive health.
Technical Background and Challenges
Probe Chemistry Considerations
The Illumina Infinium Methylation BeadChips utilize two distinct probe chemistries with different technical characteristics that must be considered during normalization. Infinium I probes employ two separate probes per CpG site—one for the methylated state and one for the unmethylated state—with the color channel determined by the nucleotide adjacent to the target cytosine (Cy3 for G/C and Cy5 for A/T) [71]. In contrast, Infinium II probes use a single probe with a color-discriminating single-base extension to distinguish methylation states, making them more economical but confounding color channel with methylation measurement [71]. Critically, Infinium II probes demonstrate a reduced dynamic range of measured methylation values compared to Infinium I probes, presumably due to using a single bead where methylated and unmethylated signals become prone to residual emission by the other dye [71]. This technical disparity creates systematic biases that normalization must address, particularly for sperm epigenetics where certain genomic regions show characteristically different methylation patterns compared to somatic tissues [37].
Sperm-Specific Considerations
Sperm cells exhibit unique methylation patterns that complicate data normalization. Research has shown that sperm DNA methylation patterns are remarkably stable yet distinct from somatic tissues, with age-related changes demonstrating predominantly demethylation trends in promoter regions [37]. One study analyzing semen-derived DNA samples found that age-related demethylation occurs inside gene regions more frequently than expected, characterizing 60.6% of significantly age-correlated differentially methylated sites [37]. When designing normalization strategies for sperm epigenetics, researchers must consider that conventional approaches optimized for blood or other somatic tissues may not directly translate to semen samples. Furthermore, sperm studies often involve compromised DNA typical of forensic semen stains, which may be of low quality and quantity, further exacerbated by the bisulfite conversion process [37]. These factors necessitate specialized normalization approaches that account for both the technical artifacts of the platform and the biological uniqueness of sperm methylation patterns.
Normalization Methodologies
Preprocessing and Quality Control
Robust quality control measures are essential prerequisites before normalization. The DRAGEN Array Methylation QC software provides high-throughput, quantitative reporting of 21 control metrics for Infinium Methylation microarrays, including detection p-values and proportion of passing assays [9]. For sperm samples specifically, additional verification of somatic cell contamination should be performed using markers like the DLK1 locus, which is highly methylated in somatic cells but essentially unmethylated in sperm cells [3]. Following quality assessment, background correction should be applied using the out-of-band channel signal from Infinium-I probes, which can be co-opted for parameterizing background subtraction [4]. The methylated and unmethylated signal intensities should undergo background correction before any normalization, as the raw fluorescence signals contain background noise from non-specific hybridization and fluorescence emissions [71].
Table 1: Quality Control Metrics for Sperm Methylation Studies
| QC Metric | Target Value | Assessment Method | Sperm-Specific Considerations |
|---|---|---|---|
| Bisulfite Conversion Efficiency | >99.5% | Control probe intensities | Critical for semen samples with degraded DNA |
| Detection P-value | <0.01 | Proportion of significantly detected probes | Filter poorly performing probes |
| Somatic Contamination | DLK1 methylation <5% | DLK1 locus analysis | Ensure pure sperm DNA population |
| Sample Sex Consistency | XY pattern | X/Y chromosome probes | Confirm male origin of samples |
| Array Intensity | >50% of probes above background | Signal intensity distribution | May be lower in forensic semen stains |
Normalization Techniques
Several normalization approaches have been developed specifically for Infinium Methylation data, each with distinct advantages and limitations for sperm epigenetics research:
Within-array normalization methods address technical differences between probe types. Peak-based correction (PBC) adjusts the distribution of Infinium II probes to match that of Infinium I probes, mitigating the reduced dynamic range of Infinium II chemistry [71]. Subset quantile normalization (SQN) performs quantile normalization separately for Infinium I and II probes, then aligns the distributions, preserving biological variability while removing technical bias [9]. For sperm studies, where global hypomethylation is common, these methods help maintain accurate quantification across different methylation density regions.
Between-array normalization corrects for technical variation across different samples processed in separate batches. Quantile normalization remains widely used, though it assumes nearly identical methylation distributions across samples—an assumption that may not hold for sperm studies comparing different fertility statuses or age groups [71]. Beta-mixture quantile normalization (BMIQ) provides a more sophisticated approach by estimating a mixture of beta distributions to model the different methylation states (hypomethylated, hemimethylated, and hypermethylated) and performing quantile normalization within each state [9]. This is particularly valuable for sperm epigenetics, where specific genomic regions may show distinct methylation patterns related to fertility status.
Batch-effect correction is crucial when samples are processed across multiple slides or different time points. The ComBat and Harman software packages effectively remove batch-effects associated with processing day, individual glass slide, and array position [71]. These methods should be applied with caution, as they may mistake biological variance for technical variance if confounding exists between batches and experimental groups. For sperm epigenetic aging studies, where subtle methylation changes are expected, it's essential to preserve true biological signals while removing technical artifacts [37].
Normalization workflow for sperm methylation data
Implementation Protocols
Comprehensive Normalization Protocol for Sperm Samples
This step-by-step protocol describes the complete normalization procedure for Infinium Methylation BeadChip data from sperm samples:
Step 1: Data Import and Quality Control
- Import raw IDAT files using the
minfiorSeSAMepackages in R [9]. - Calculate detection p-values and filter probes with p > 0.01 in more than 5% of samples.
- Verify bisulfite conversion efficiency using built-in control probes.
- For sperm-specific QC, assess potential somatic cell contamination using the DLK1 locus or similar markers [3].
Step 2: Background Correction
- Apply background correction using the
preprocessNoobmethod inminfior similar functionality inSeSAMe, which utilizes the out-of-band signals from Infinium I probes [9]. - This step subtracts background fluorescence using a normal-exponential convolution model to estimate and remove technical noise.
Step 3: Within-Array Normalization
- Implement subset quantile normalization (SQN) using the
preprocessSqnfunction inminfior similar approaches inSeSAMe. - This method normalizes Infinium I and II probes separately then aligns their distributions.
- Alternatively, apply peak-based correction (PBC) for studies focusing on epigenetic age prediction in sperm [37].
Step 4: Between-Array Normalization
- Perform quantile normalization across all samples using
preprocessQuantileinminfior equivalent functions. - For sperm studies with expected global methylation differences, consider using Beta-Mixture Quantile Dilution (BMIQ) normalization to preserve biological variability.
Step 5: Batch-Effect Correction
- Identify batch variables (processing date, slide, position) and biological covariates (age, BMI) [3].
- Apply ComBat from the
svapackage, specifying biological covariates to preserve. - Validate correction using PCA visualization before and after adjustment.
Step 6: Probe Filtering
- Remove probes containing SNPs at the CpG site or single-base extension.
- Eliminate cross-reactive probes that map to multiple genomic locations.
- Filter sex chromosome probes if analyzing autosomal methylation only.
- For EPICv2 arrays, utilize updated annotation packages that provide improved probe mapping information [4] [72].
Specialized Normalization for Sperm Epigenetic Aging
Research on epigenetic age prediction in semen requires specialized normalization approaches. Studies have identified numerous differentially methylated sites in semen that continuously change over an individual's lifetime, with most age-correlated sites showing demethylation trends [37]. When normalizing data for epigenetic age prediction:
- Preserve subtle, continuous methylation changes at age-correlated CpG sites like those in SH2B2, EXOC3, IFITM2, GALR2, and FOLH1B genes [37].
- Avoid over-correction that might remove true biological aging signals, which can be particularly challenging given the different aging patterns in sperm compared to somatic tissues [37].
- Consider including known covariates like BMI in the normalization model, as research suggests BMI may have a subtle association with epigenetic age acceleration in sperm, though this relationship requires further validation [3].
Table 2: Normalization Methods Comparison for Sperm Epigenetics
| Method | Primary Use | Advantages | Limitations for Sperm Research |
|---|---|---|---|
| PreprocessNoob | Background correction | Utilizes out-of-band probes | May over-correct low signal semen samples |
| Subset Quantile | Within-array normalization | Handles probe type differences | Assumes similar distribution across types |
| BMIQ | Between-array normalization | Models methylation states | Complex implementation |
| ComBat | Batch-effect correction | Preserves biological variables | Risk of removing true biological signals |
| Peak-Based Correction | Probe-type adjustment | Maintains dynamic range | Less effective for globally hypomethylated samples |
The Scientist's Toolkit
Essential Research Reagents and Software
Table 3: Essential Research Reagents and Computational Tools
| Item | Function | Application in Sperm Epigenetics |
|---|---|---|
| Infinium MethylationEPIC v2.0 BeadChip | Genome-wide methylation profiling | Interrogates >935,000 CpG sites; improved coverage of enhancer regions relevant to spermatogenesis [4] |
| Bisulfite Conversion Kit | Converts unmethylated C to U | Critical step for methylation detection; efficiency crucial for semen samples with degraded DNA [37] |
| DNA Extraction Kit (sperm-specific) | Isulates DNA from semen | Protocols optimized for sperm cell lysis and removal of somatic contamination [3] |
| SeSAMe R Package | End-to-end data analysis | Implements improved normalization techniques specifically for Infinium arrays [9] |
| Minfi R Package | Comprehensive methylation analysis | Provides multiple preprocessing and normalization methods; widely used in epigenetic studies [9] |
| IlluminaHumanMethylationEPICv2anno.20a1.hg38 | Probe annotation | Updated annotations for EPICv2 arrays with improved probe mapping [72] |
| ChAMP Package | Epigenome-Wide Analysis | Integrated pipeline for normalization, DMP detection, and enrichment analysis [9] |
Tool relationships in sperm methylation analysis
Troubleshooting and Validation
Addressing Common Normalization Issues
Several challenges commonly arise when normalizing sperm methylation data, along with specific solutions:
Problem: Poor normalization due to global hypomethylation
- Solution: Implement stratified normalization that accounts for the characteristic hypomethylation pattern of sperm cells. Use BMIQ normalization that models different methylation states rather than assuming a uniform distribution across samples.
Problem: Batch effects correlated with experimental groups
- Solution: Utilize Harman correction instead of ComBat when batch is completely confounded with experimental groups, as it uses a probabilistic approach to separate technical and biological variance [71].
Problem: Inaccurate age prediction due to over-normalization
- Solution: Validate normalization by assessing known age-correlated CpG sites in sperm (e.g., sites in TUBB3, EXOC3, TBX4) [37]. The correlation structure should be preserved post-normalization.
Problem: Low signal intensity from compromised semen DNA
- Solution: Apply background correction methods specifically designed for low-quality samples, such as the
preprocessFunnormavailable inminfi, which includes functional normalization adapted for degraded samples [37].
Validation Metrics for Sperm Epigenetics
After normalization, several validation steps should be performed to ensure successful technical artifact removal without eliminating biological signals:
- Technical replication correlation: Assess correlation between technical replicates, which should show high concordance (Spearman's rho >0.98) [4].
- Sex chromosome verification: Confirm that X and Y chromosome probes show expected patterns for male samples.
- Sperm-specific markers: Verify that known sperm-specific methylation patterns are maintained (e.g., hypomethylation at imprinted loci).
- Epigenetic age prediction: Apply established sperm epigenetic age models to a subset of samples with known age; the mean absolute error should approximate established values (~5.1 years) [37].
- Batch-effect assessment: Perform principal component analysis to confirm removal of batch-associated variance while preserving biological variance.
By implementing these comprehensive normalization and background correction strategies, researchers can significantly enhance data quality for sperm epigenetics studies using Infinium Methylation BeadChips, leading to more reliable detection of biological phenomena related to male fertility, epigenetic inheritance, and reproductive health.
Benchmarking Performance: EPICv2 Improvements and Cross-Platform Concordance
Evaluating Technical Reproducibility and Sensitivity in Sperm Samples
The application of Infinium Methylation BeadChip technology in sperm epigenetics research offers unprecedented opportunities to uncover paternal influences on offspring development and complex disease etiology. However, the technical reproducibility and sensitivity of methylation analyses in sperm samples present unique challenges that must be systematically addressed to generate reliable, interpretable data. Sperm cells possess distinct epigenetic landscapes compared to somatic cells, with widespread erasure and re-establishment of methylation patterns during gametogenesis, making them particularly susceptible to technical artifacts and biological contamination [2]. This application note provides a comprehensive framework for evaluating and ensuring technical reproducibility and sensitivity when utilizing Infinium Methylation BeadChip platforms for sperm epigenetics research, with specific protocols designed to address the unique challenges of working with spermatozoa.
The fundamental challenge in sperm methylation studies stems from two primary sources: the inherent biological variability of semen parameters and the persistent risk of somatic cell contamination. Semen analysis parameters demonstrate significant within-subject variability, with coefficients of variation ranging from 36% for volume and motility to 82% for total motile count [73]. This biological variability directly impacts the epigenetic assessment, as differential methylation patterns may reflect technical rather than biological phenomena. Furthermore, somatic DNA contamination in semen samples can severely compromise data interpretation, as somatic methylation patterns differ dramatically from those in spermatozoa [2]. Without rigorous quality control measures, researchers risk drawing misleading conclusions about sperm-specific epigenetic signatures.
Quantitative Assessment of Technical Variability in Sperm Analyses
Understanding the sources of variation in sperm epigenetic analyses requires consideration of both pre-analytical and analytical factors. The inherent variability of semen parameters establishes a baseline for expected technical variability in subsequent epigenetic assessments. Table 1 summarizes the reproducibility metrics for conventional semen parameters, which directly inform expectations for epigenetic analyses.
Table 1: Reproducibility of Semen Analysis Parameters in Youths at Risk for Infertility
| Semen Parameter | Within-Subject Coefficient of Variation (CVw) | Intraclass Correlation Coefficient (ICC) | Concordance Rate (%) |
|---|---|---|---|
| Volume | 36% | 0.78 [0.67–0.85] | 86% |
| Density | 64% | 0.84 [0.76–0.90] | 81% |
| Total Count | 72% | 0.88 [0.82–0.92] | 92% |
| Motility | 36% | 0.55 [0.39–0.68] | 77% |
| Total Motile Count | 82% | 0.78 [0.67–0.85] | 85% |
Data adapted from [73]
The data demonstrate that while certain parameters like total count show high reliability (ICC = 0.88), they also exhibit substantial within-subject variability (CVw = 72%). This paradox highlights the necessity of replicate sampling and appropriate statistical modeling when designing sperm epigenetics studies. The total motile count, often considered the most clinically relevant parameter for fertility assessment, shows the highest degree of variability (CVw = 82%), suggesting that studies correlating methylation patterns with this parameter require particularly robust sample sizes and replication strategies [73].
Impact of Somatic Contamination on Methylation Measurements
Somatic cell contamination represents a particularly insidious source of technical artifacts in sperm methylation studies. Unlike biological variability, which can be accounted for statistically, contamination introduces systematic errors that can completely obscure true sperm-specific methylation patterns. Research has identified 9,564 CpG sites that serve as effective markers for detecting somatic DNA contamination, with these sites being highly methylated in blood samples compared to sperm but unrelated to infertility status [2]. The magnitude of this effect necessitates rigorous quality control, with recommendations to apply a 15% cutoff during data analysis to completely eliminate the influence of somatic DNA contamination in sperm epigenetic studies [2].
Comprehensive Experimental Protocols
Protocol 1: Sperm Sample Collection and Purification
Principle: To obtain pure sperm populations free from somatic cell contamination while preserving epigenetic integrity.
Reagents and Materials:
- Somatic Cell Lysis Buffer (SCLB): 0.1% SDS, 0.5% Triton X-100 in distilled water
- Density gradient centrifugation media (e.g., Percoll or Sil-Select)
- Phosphate-buffered saline (PBS)
- DNase/RNase-free water
- Phase-contrast microscope
- Refrigerated centrifuge
Procedure:
- Collect semen samples after a recommended abstinence period of 3-5 days to minimize biological variability [73].
- Allow samples to liquefy completely at 37°C for 20-30 minutes.
- Perform initial microscopic examination to assess sperm concentration, motility, and presence of round cells (potential somatic contamination).
- For somatic cell removal, mix 1 mL of semen with 2 mL of Somatic Cell Lysis Buffer, incubate at 4°C for 5 minutes, then centrifuge at 500 × g for 10 minutes [2].
- Carefully remove supernatant and resuspend pellet in 1 mL PBS.
- Layer the sample onto a density gradient and centrifuge at 300 × g for 20 minutes.
- Collect the sperm-rich fraction from the bottom of the gradient and wash twice with PBS.
- Verify purity by microscopic examination, with specific attention to the absence of round cells.
- Aliquot purified sperm samples and store at -80°C until DNA extraction.
Quality Control Measures:
- Purity Assessment: Examine under microscope after purification; somatic cells should be <1% of total cells.
- Molecular Purity Check: Analyze methylation status of the DLK1 locus, which is highly methylated in somatic cells but unmethylated in pure sperm samples [3].
- Contamination Marker Screening: Assess a panel of 9,564 CpG sites known to differentiate between sperm and blood methylation patterns [2].
Protocol 2: DNA Extraction and Bisulfite Conversion for BeadChip Analysis
Principle: To extract high-quality DNA from sperm samples and complete efficient bisulfite conversion compatible with Infinium BeadChip analysis.
Reagents and Materials:
- Sperm-specific DNA extraction kit (e.g., with additional reducing agents to break disulfide bonds)
- Commercial bisulfite conversion kit with >99% conversion efficiency
- Agarose gel electrophoresis system
- Spectrophotometer (NanoDrop) or fluorometer (Qubit)
- Thermal cycler
Procedure:
- Extract DNA using a sperm-specific protocol that includes dithiothreitol (DTT) or similar reducing agents to efficiently break down sperm chromatin packaging.
- Quantify DNA concentration using fluorometric methods (e.g., Qubit) for superior accuracy with bisulfite-converted DNA.
- Assess DNA quality by agarose gel electrophoresis to confirm high molecular weight and absence of degradation.
- Perform bisulfite conversion using a commercial kit optimized for maximum conversion efficiency (>99%).
- Use 500 ng input DNA as recommended for most kits
- Follow manufacturer's protocol for incubation conditions
- Include unconverted control DNA to assess conversion efficiency
- Purify bisulfite-converted DNA and elute in appropriate buffer for BeadChip application.
- Verify conversion efficiency by including control CpG sites known to be uniformly methylated or unmethylated in sperm.
Troubleshooting:
- Low DNA yield: Increase DTT concentration or incubation time during extraction
- Incomplete bisulfite conversion: Ensure fresh bisulfite reagents and proper pH conditions
- DNA degradation: Reduce freeze-thaw cycles and include nuclease inhibitors during extraction
Protocol 3: Infinium BeadChip Processing and Quality Assessment
Principle: To generate high-quality methylation data from sperm samples using Infinium BeadChip technology with appropriate quality control metrics.
Reagents and Materials:
- Infinium MethylationEPIC BeadChip Kit or equivalent
- BeadArray scanner
- GenomeStudio Software or alternative analysis pipeline (e.g., SeSAMe, Minfi)
- High-quality reference DNA for controls
Procedure:
- Process bisulfite-converted DNA according to manufacturer's protocol for the appropriate Infinium BeadChip platform.
- Hybridize samples to BeadChips using standardized conditions.
- Scan BeadChips using appropriate laser settings and generate intensity data files.
- Import data into analysis software (e.g., GenomeStudio, R-based pipelines like SeSAMe or Minfi).
- Perform initial quality assessment using the following criteria:
- Detection p-values < 0.01 for all samples
- Average intensity values > 4,000
- Bisulfite conversion controls indicating >99% efficiency
- Staining, extension, and hybridization controls within expected ranges
- Apply normalization procedures appropriate for sperm samples (e.g., subset-quantile within array normalization [SWAN] or beta-mixture quantile normalization [BMIQ]).
- Export beta-values and M-values for downstream analysis.
Quality Assessment Metrics:
- Sample-dependent controls: Evaluate hybridization, target removal, extension, and staining efficiency
- Bisulfite conversion efficiency: Assess using built-in control probes
- Signal intensity thresholds: Remove probes with detection p-value > 0.01
- Contamination screening: Apply the 9,564 CpG somatic contamination panel and remove samples exceeding 15% contamination threshold [2]
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 2: Key Research Reagent Solutions for Sperm Methylation Studies
| Reagent/Solution | Function | Application Notes |
|---|---|---|
| Somatic Cell Lysis Buffer (SCLB) | Selective lysis of non-sperm cells | Critical for removing leukocytes and other contaminating somatic cells; composition: 0.1% SDS, 0.5% Triton X-100 [2] |
| Dithiothreitol (DTT) | Reduction of sperm protamine disulfide bonds | Essential for efficient DNA extraction from tightly packaged sperm chromatin; typically used at 5-10 mM concentration |
| Density Gradient Media (Percoll/Sil-Select) | Sperm purification based on motility and morphology | Separates motile, morphologically normal sperm from immotile sperm and cellular debris |
| Bisulfite Conversion Kit | Chemical conversion of unmethylated cytosines to uracils | Required for methylation detection; select kits with >99% conversion efficiency for reliable results [74] |
| DNA Methylation Standards | Controls for methylation quantification | Include fully methylated and unmethylated DNA standards for assay calibration |
| Somatic Contamination Marker Panel | Detection of non-sperm DNA contamination | 9,564 CpG sites identified as highly methylated in blood vs. sperm; critical for quality assessment [2] |
| DLK1 Locus Control | Verification of sperm sample purity | Locus highly methylated in somatic cells but unmethylated in sperm; qualitative purity assessment [3] |
Data Analysis Framework for Technical Reproducibility
Quality Control Pipeline Implementation
A robust quality control pipeline is essential for establishing technical reproducibility in sperm methylation studies. The following workflow represents the critical steps for ensuring data quality:
Statistical Assessment of Technical Reproducibility
To quantitatively evaluate technical reproducibility, researchers should implement the following statistical measures:
Intraclass Correlation Coefficient (ICC): Calculate ICC for replicate samples to assess reliability of methylation measurements. Benchmarks from semen analysis parameters provide context, with ICC > 0.8 representing almost perfect agreement [73].
Coefficient of Variation (CV): Determine within-subject CV for technical replicates. Based on semen parameter variability, CV < 40% may represent acceptable technical variation for sperm methylation analyses [73].
Concordance Rates: Establish agreement rates between replicate measurements for dichotomized methylation status (hyper/hypomethylated). Target concordance rates >85% based on semen parameter benchmarks [73].
Differential Methylation Analysis: When comparing experimental groups, apply strict multiple testing correction (e.g., Bonferroni or False Discovery Rate) and effect size thresholds to minimize false positives. Include covariates for known technical factors (e.g., batch effects, sample purity) in statistical models.
Validation Strategies for Methylation Findings
Technical Validation Approaches
Given the technical challenges specific to sperm methylation analyses, independent validation of significant findings is essential. Table 3 compares common validation methods for DNA methylation results.
Table 3: Comparison of DNA Methylation Validation Methods
| Method | Principle | Accuracy | Throughput | Key Applications | Limitations |
|---|---|---|---|---|---|
| Pyrosequencing | Sequencing by synthesis with quantitative light detection | High (quantitative) | Medium | Validation of individual CpG sites; suitable for CpG-poor and CpG-rich regions | Limited to short reads (~100 bp); instrument cost [74] |
| MS-HRM | High-resolution melting analysis of bisulfite-converted DNA | High | High | Rapid screening of methylation patterns; cost-effective for large sample sets | Limited quantitative precision for intermediate methylation [74] |
| Targeted Bisulfite Sequencing | Deep sequencing of bisulfite-converted target regions | Very high | Medium-high | Gold standard for comprehensive regional methylation assessment | Higher cost than other targeted methods [75] |
| MSRE-qPCR | Methylation-sensitive restriction digestion followed by qPCR | Medium | High | Rapid assessment of specific restriction sites; no bisulfite conversion required | Limited to enzymes' recognition sites; not single-CpG resolution [74] |
Biological Validation Considerations
For sperm methylation studies, biological validation should include:
- Correlation with functional outcomes (e.g., sperm motility, fertilization rates)
- Stability assessment across multiple collections from the same individual
- Integration with other molecular data (e.g., chromatin accessibility, transcriptomics)
- Cross-validation in independent cohorts when possible
Ensuring technical reproducibility and sensitivity in sperm methylation studies using Infinium BeadChip technology requires a comprehensive approach addressing both pre-analytical and analytical variables. Based on current evidence, the following recommendations emerge as critical for robust sperm epigenetics research:
Implement Rigorous Somatic Cell Removal: Combine mechanical separation (density gradient centrifugation) with chemical lysis (SCLB treatment) followed by molecular verification using the 9,564 CpG contamination panel with a strict 15% cutoff threshold [2].
Account for Biological Variability: Collect multiple samples per individual when possible, recognizing the high within-subject variability in semen parameters (CVw 36-82%) [73]. Power studies should incorporate this variability into sample size calculations.
Apply Sperm-Specific Analytical Methods: Utilize analysis pipelines (e.g., SeSAMe, Minfi) with parameters optimized for sperm-specific methylation patterns, including appropriate normalization methods and contamination screening [9].
Implement Multiplex Validation Strategies: Combine high-throughput screening with targeted validation using orthogonal methods such as pyrosequencing or targeted bisulfite sequencing for confirmed findings [74] [75].
Standardize Reporting: Document all quality control metrics, including purity assessments, bisulfite conversion efficiency, detection rates, and contamination screening results to enable proper evaluation of technical reproducibility.
As sperm epigenetics continues to evolve as a field, maintaining rigorous standards for technical reproducibility and sensitivity will be paramount for generating biologically meaningful insights into paternal epigenetic contributions to development and disease.
The Infinium MethylationEPIC v2.0 BeadChip (EPICv2) represents a significant evolution in microarray technology for DNA methylation analysis. With 936,866 total probes, it covers over 930,000 unique methylation sites, providing extensive genome-wide coverage at a cost-effective price point, making it ideal for large-scale epigenome-wide association studies (EWAS) [25]. This updated version retains a substantial portion of content from its predecessor, the MethylationEPIC v1.0 (EPICv1), while substantially expanding coverage in biologically significant genomic regions.
Probe Content and Design Improvements
EPICv2 incorporates several critical design enhancements that improve data quality and utility across diverse research applications, including sperm epigenetics:
- Backward Compatibility: EPICv2 retains approximately 77% of probes from EPICv1, ensuring continuity with existing data and tools [55] [76]. This includes 721,378 shared probes that enable direct comparison between versions [77].
- Expanded Regulatory Coverage: The array adds over 200,000 new probes specifically targeting enhancers, open chromatin regions, and CTCF-binding domains, providing enhanced coverage of regulatory elements [55] [76].
- Probe Optimization: Approximately 143,000 poorly performing probes from EPICv1 have been removed, with about 73% of these being probes susceptible to interference from underlying sequence polymorphisms [55] [76].
- Specialized Content: EPICv2 introduces 824 "nv" probes targeting recurrent somatic mutations in cancer, expanding its utility for cancer epigenetics research [4].
Probe Type Distribution
| Probe Category | EPICv2 Count | Comparison to EPICv1 |
|---|---|---|
| Total Probes | 936,866 [55] / 937,690 [4] | ~77% of EPICv1 probes retained |
| CpG Methylation ("cg" probes) | >99% of total [4] | 83% of EPICv1 cg probes retained |
| Non-CpG Methylation ("ch" probes) | Comparable to previous arrays [4] | Similar to EPICv1 |
| SNP Probes ("rs" probes) | Comparable to previous arrays [4] | 57 SNP probes available for both versions [77] |
| Somatic Mutation ("nv" probes) | 824 new probes [4] | Not present in EPICv1 |
| Control Probes | Comparable to previous arrays [4] | Similar to EPICv1 |
Enhanced Probe Mapping and Ancestry Diversity
Improved Genomic Alignment and Specificity
EPICv2 demonstrates substantial improvements in probe mapping accuracy and specificity, critical factors for reliable methylation quantification in diverse populations:
- GRCh38/h38 Alignment: EPICv2 features annotation to the most recent GRCh38/h38 human genome build, resulting in fewer poorly mapping probes compared to EPICv1 [55] [4].
- Reduced Cross-Reactivity: The removal of problematic probes from EPICv1 and optimized new probe designs minimize cross-hybridization potential, enhancing measurement specificity [4].
- Strand Information: EPICv2 incorporates explicit strand designation in probe naming conventions (Watson or Crick strand), improving annotation clarity [4].
Performance Across Diverse Ancestries
The enhanced probe design of EPICv2 specifically addresses limitations in diverse population studies:
- Reduced Ancestry-Specific Bias: Fewer probes are subject to direct influence by ancestry-specific genetic variation, though African ancestry populations still show slightly higher susceptibility due to greater genetic diversity [4].
- Improved Population Compatibility: Optimized probe selection minimizes the impact of underlying sequence polymorphisms, making the array more reliable across diverse human populations [55] [76].
Technical Performance and Reproducibility
EPICv2 demonstrates excellent technical performance characteristics:
- High Reproducibility: Technical replicates show high correlation coefficients (Spearman's rho), significantly exceeding correlations between different cell lines [4].
- Low-Input Compatibility: The array supports DNA input down to 1 nanogram, facilitating studies with limited starting material [4].
- Sample Compatibility: Compatible with both blood samples and FFPE tissue samples, enabling utilization of large biorepositories [25].
Figure 1: EPICv2 Probe Content Evolution and Key Improvements. The diagram illustrates the transformation from EPICv1 to EPICv2 content, highlighting the removal of problematic probes, retention of high-quality content, and addition of new biologically relevant probes, culminating in improved technical characteristics.
Experimental Protocols for Sperm Epigenetics Research
DNA Extraction and Bisulfite Conversion Protocol for Sperm Samples
Principle: Sperm DNA extraction requires specialized approaches to account for unique chromatin organization, including high protamine content and disulfide bond cross-linking.
Reagents and Equipment:
- Lysis Buffer (Tris-HCl, EDTA, SDS, DTT)
- Proteinase K
- Phenol:Chloroform:Isoamyl Alcohol (25:24:1)
- Zymo Research Bisulfite Conversion Kit or equivalent
- 100% Ethanol and 70% Ethanol
- Nuclease-free Water
- Thermal Cycler
- Microcentrifuge
- Spectrophotometer/Nanodrop
Procedure:
- Sperm Preparation and Lysis:
- Isolate sperm from semen samples using density gradient centrifugation.
- Resuspend sperm pellet in 500μL lysis buffer with 20μL 1M DTT and 20μL Proteinase K (20mg/mL).
- Incubate at 56°C for 2-4 hours with occasional mixing until completely lysed.
DNA Extraction:
- Add equal volume phenol:chloroform:isoamyl alcohol, mix thoroughly, and centrifuge at 14,000×g for 5 minutes.
- Transfer aqueous phase to new tube and add 2 volumes of 100% ethanol to precipitate DNA.
- Centrifuge at 14,000×g for 10 minutes, wash pellet with 70% ethanol, and air dry.
- Resuspend DNA in nuclease-free water and quantify using spectrophotometry.
Bisulfite Conversion:
- Use 250-500ng DNA input for bisulfite conversion following manufacturer protocols.
- Program thermal cycler: Denaturation at 95°C for 30 seconds, incubation at 50°C for 60 minutes (repeat 10-20 cycles), hold at 4°C.
- Purify bisulfite-converted DNA and elute in appropriate buffer.
Quality Control:
- Verify DNA concentration and purity (A260/280 ratio 1.8-2.0).
- Confirm complete bisulfite conversion using control reactions.
- Store converted DNA at -80°C if not used immediately.
EPICv2 Array Processing and Quality Assessment
Principle: The Infinium HD Methylation Assay utilizes bead-bound oligonucleotides to query methylation status at single-CpG-site resolution after bisulfite conversion.
Reagents and Equipment:
- Infinium MethylationEPIC v2.0 Kit
- iScan System or NextSeq 550 System
- BeadChip Hyb Chambers
- Illumina GenomeStudio Software or equivalent
Procedure:
- Whole Genome Amplification:
- Amplify 250ng bisulfite-converted DNA using kit reagents.
- Fragment amplified DNA enzymatically.
BeadChip Hybridization:
- Apply fragmented DNA to EPICv2 BeadChip and incubate overnight (16-24 hours) for hybridization.
- Perform single-base extension with labeled nucleotides.
Staining and Imaging:
- Stain BeadChip with fluorescent labels.
- Image BeadChip using iScan scanner or equivalent.
Data Processing:
- Extract intensity data using GenomeStudio or similar software.
- Apply normalization (functional normalization recommended for version comparisons) [77].
Quality Assessment:
- Check sample-independent controls (staining, extension, hybridization).
- Verify bisulfite conversion efficiency using control probes.
- Assess sample quality metrics (detection p-values, signal intensities).
Data Analysis Considerations for Sperm Studies
Special Considerations for Sperm Epigenetics:
- Sperm DNA exhibits unique methylation patterns with global levels of approximately 70-80% in mature sperm, lower than most somatic cells [78].
- Non-CpG methylation accumulates in sperm during fetal development, particularly around B1 SINE transposon sequences [78].
- Imprinted genes escape epigenetic reprogramming and require careful analysis.
Analysis Workflow:
- Preprocessing: Address EPICv2-specific features including:
- 5,100 replicate probes with 2-10 replicates each, differentiated by name and sequence [55]
- Probe filtering based on detection p-values
- Normalization to address technical variation
- Differential Methylation Analysis:
- Account for EPIC version effects when comparing with historical data
- Focus on biologically relevant regions for sperm function
- Consider cell-type heterogeneity in sperm samples
Figure 2: EPICv2 Sperm Methylation Analysis Workflow. The diagram outlines the complete experimental process from sample collection through data analysis, highlighting critical steps specific to sperm epigenetics research and EPICv2-specific processing requirements.
Comparative Performance Data
Quantitative Performance Metrics
| Performance Characteristic | EPICv2 Performance | Comparison to EPICv1 |
|---|---|---|
| Technical Reproducibility | High correlation between replicates (Spearman's rho) [4] | Similar to EPICv1 performance |
| Cross-Version Concordance | High agreement at array level [55] | Variable agreement at individual probe level |
| Sample Input Requirements | Compatible with low-input DNA (1ng demonstrated) [4] | Similar input requirements (250ng standard) |
| Cell Type Discrimination | Improved discrimination for added probes [4] | Lower inter-cell line correlation for shared probes |
| Infinium Chemistry Changes | 70 Infinium-I to II switches, 12 Infinium-II to I switches [4] | Probes with altered designs show higher methylation differences |
Impact on DNA Methylation-Based Predictive Tools
EPICv2's probe changes affect various DNA methylation-based algorithms and tools commonly used in epigenetic research:
- Epigenetic Clocks: Most but not all predictive CpGs are retained on EPICv2 [55] [76]
- Cell Type Deconvolution: Algorithms show modest but significant differences between versions [77]
- Biomarker Predictors: Inflammation and lifestyle biomarkers require version-specific adjustment [55]
Recommendation: When harmonizing data across EPIC versions, apply statistical adjustments for EPIC version or calculate estimates separately for each version to mitigate version-specific discordances [55] [77] [76].
The Scientist's Toolkit: Essential Research Reagents and Materials
| Research Reagent | Function/Application | Specifications/Notes |
|---|---|---|
| Infinium MethylationEPIC v2.0 Kit | Genome-wide methylation profiling | 8 samples per array; requires 250ng DNA input; includes all reagents except bisulfite conversion kits [25] |
| Zymo Research Bisulfite Conversion Kit | DNA bisulfite conversion | Compatible with EPICv2; required for cytosine-to-uracil conversion of unmethylated cytosines |
| DTT (Dithiothreitol) | Sperm chromatin disruption | Reduces disulfide bonds in protamine-DNA complexes for efficient DNA extraction |
| Proteinase K | Protein digestion | Facilitates sperm cell lysis and DNA release |
| Phenol:Chloroform:Isoamyl Alcohol | DNA purification | Separates DNA from proteins and other cellular components |
| Flowsorted.Blood.EPIC R Package | Cell type deconvolution | Reference-based estimation of white blood cell composition [79] |
| MethylCallR R Package | Data analysis pipeline | Controls duplicated probes in EPICv2; enables conversion between array versions [79] |
Within the specialized field of sperm epigenetics research, selecting an appropriate DNA methylation profiling technology is paramount. The Infinium Methylation BeadChip has been a cornerstone for many large-scale epigenetic studies due to its user-friendly data analysis and high-throughput capability [4]. However, for investigations focused on specific candidate regions or requiring higher sample throughput, targeted bisulfite sequencing (BS) presents a potentially reliable and cost-effective alternative [80]. This application note evaluates the concordance between these two platforms and provides detailed protocols for implementing targeted bisulfite sequencing in the context of sperm epigenetics research, addressing unique challenges such as somatic cell contamination.
Performance Comparison: BeadChip vs. Bisulfite Sequencing
Quantitative Concordance Metrics
Direct comparisons between the Infinium MethylationEPIC BeadChip and various bisulfite sequencing methods demonstrate strong technical agreement, validating BS as a viable alternative for methylation profiling.
Table 1: Cross-Platform Concordance in DNA Methylation Profiling
| Comparison | Correlation (R²) | Sample Type | Key Finding |
|---|---|---|---|
| Targeted BS vs. EPIC Array [80] | High sample-wise correlation | Ovarian tissue, Cervical swabs | Strong correlation, especially in tissue samples; slightly lower in swabs due to DNA quality. |
| TMS (EM-seq) vs. EPIC Array [81] | 0.97 | Human DNA | Enzymatic methylation sequencing shows very strong agreement with the array. |
| TMS (EM-seq) vs. WGBS [81] | 0.99 | Human DNA | Optimized targeted methods achieve near-perfect agreement with the gold standard. |
| TMS vs. RRBS [81] | 0.98 | Non-human primates | High concordance in cross-species applications. |
Sperm Epigenetics: Specific Considerations and Somatic Contamination
Sperm cells possess a unique epigenome distinct from somatic cells, making pure sperm isolation critical for accurate analysis. Somatic DNA contamination in semen samples, particularly problematic in oligozoospermic individuals, can significantly skew methylation results [20]. A robust strategy to address this includes:
- Microscopic Examination: Initial visual inspection to detect somatic cells.
- Somatic Cell Lysis Buffer (SCLB) Treatment: Incubation with a buffer containing 0.1% SDS and 0.5% Triton X-100 to lyse contaminating cells [20].
- Biomarker Verification: Using a panel of 9,564 identified CpG sites that are highly methylated in blood (>80%) but minimally methylated in sperm (<20%) to detect residual contamination [20].
- Data Analysis Cut-off: Applying a 15% threshold during differential methylation analysis to eliminate confounding effects from low-level undetected contamination [20].
Experimental Protocols
Protocol 1: Custom Targeted Bisulfite Sequencing Panel
This protocol is adapted from a study comparing a custom BS panel to the Infinium MethylationEPIC array [80] and is suitable for analyzing sperm DNA.
Workflow Overview
Step-by-Step Procedure
DNA Extraction and Bisulfite Conversion
Custom Panel Design
- Design a custom panel targeting regions of interest (e.g., 648 CpG sites across 119 primers) [80].
- Include internal targets (CpGs with known diagnostic potential) and external targets (literature-based cancer-related or age-related regions) [80] [37].
- Use software like Methyl Primer Express v1.0 for oligonucleotide design [82].
Library Preparation and Amplification
- Prepare libraries using the QIAseq Targeted Methyl Custom Panel kit (QIAGEN, Cat. No. 335 602) according to the manufacturer's protocol [80].
- Perform target amplification via PCR. For longer promoter regions (>1 kb), use long PCR and nested techniques with primers containing universal tail sequences for downstream sequencing [82].
Library Quality Control and Quantification
- Assess library concentration using the QIAseq Library Quant Assay Kit (QIAGEN) [80].
- Evaluate library size distribution and quality using a Bioanalyzer High Sensitivity DNA Kit (Agilent Technologies) [80].
- For over-amplified libraries, perform reconditioning with kits like the GeneRead DNA Library Prep I Kit [80].
Sequencing
- Pool libraries in equimolar concentrations.
- Sequence on an Illumina MiSeq instrument using a v2 Reagent Kit (300 cycles), spiking in PhiX for quality control [80].
Bioinformatic Analysis
- Import raw sequencing data (.fastq) into an analysis platform such as QIAGEN CLC Genomics Workbench (v. 23.0.5) [80].
- Execute a custom workflow for alignment, methylation calling, and output of methylation levels (beta values) for each CpG site.
- Apply quality control filters: exclude samples with coverage <30x in more than 1/3 of CpG sites, and remove CpG sites with <30x coverage in over 50% of samples [80].
Protocol 2: Enzymatic Methyl-Seq (EM-seq) for Targeted Methylation Sequencing
This protocol uses enzymatic conversion instead of bisulfite, minimizing DNA damage and yielding higher-quality data [81].
Workflow Overview
Step-by-Step Procedure
DNA Input and Enzymatic Conversion
Library Preparation and Sequencing
Data Analysis
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for Targeted Bisulfite Sequencing in Sperm Epigenetics
| Product Name | Supplier | Function | Considerations for Sperm Research |
|---|---|---|---|
| QIAseq Targeted Methyl Custom Panel | QIAGEN | Library preparation for targeted BS | Enables simultaneous testing of custom targets across many samples. |
| EpiTect Bisulfite Kit / EZ DNA Methylation Kit | Zymo Research | Bisulfite conversion of DNA | Critical step for BS-based methods; requires optimized input DNA quality. |
| QIAseq Library Quant Assay Kit | QIAGEN | Library quantification | Essential for accurate pooling before sequencing. |
| Bioanalyzer High Sensitivity DNA Kit | Agilent Technologies | Library quality control | Assesses fragment size distribution and library integrity. |
| Somatic Cell Lysis Buffer (SCLB) | Lab-prepared | Sperm sample purification | Contains 0.1% SDS, 0.5% Triton X-100. Crucial for removing somatic cell contamination prior to DNA extraction [20]. |
| Infinium MethylationEPIC v2 BeadChip | Illumina | Reference methylation profiling | Expanded coverage; supports low-input DNA (down to 1 ng); used for validation [4]. |
Targeted bisulfite sequencing and its enzymatic successor, EM-seq, demonstrate high concordance with the Infinium Methylation BeadChip, establishing them as reliable and cost-effective alternatives for focused sperm epigenetics studies. These sequencing-based approaches offer greater flexibility for custom panel design and higher multiplexing capabilities. For researchers, the choice between platforms should be guided by the specific research question, the number of samples, the required genomic coverage, and the available budget. When employing these techniques for sperm analysis, implementing a comprehensive strategy to mitigate somatic DNA contamination is non-negotiable for obtaining biologically accurate results.
Within the context of sperm epigenetics research utilizing the Infinium Methylation BeadChip, validation of genome-wide findings is a critical step to ensure data integrity and biological relevance. High-throughput arrays, while powerful for discovery, can be influenced by technical artifacts and require confirmation via methods based on differing biochemical principles. This document outlines the application of two established orthogonal validation techniques—pyrosequencing and Comprehensive High-Throughput Arrays for Relative Methylation (CHARM)—specifically for verifying methylation signatures identified in sperm studies. The unique challenges of sperm epigenetics, particularly concerning somatic cell contamination [2], make rigorous validation paramount for drawing accurate conclusions about male fertility, environmental exposures, and transgenerational inheritance.
Pyrosequencing for Targeted DNA Methylation Validation
Pyrosequencing is a quantitative sequencing-by-synthesis method that provides precise, single-base resolution methylation levels for specific CpG sites. It is considered a gold standard for validating methylation levels obtained from BeadChip arrays due to its high accuracy, reproducibility, and sensitivity [83]. The technique relies on the sequential addition of nucleotides and the real-time detection of light released upon nucleotide incorporation into the growing DNA strand. After bisulfite conversion of DNA, the incorporation of a dATPαS (corresponding to a methylated cytosine) versus a dTTP (corresponding to an unmethylated cytosine) is quantified, and the methylation percentage is calculated from the resulting peak heights on a pyrogram [83] [84].
Advantages for Sperm Epigenetics
- High Quantitative Precision: Enables precise measurement of methylation levels at individual CpGs, crucial for detecting subtle methylation shifts that may be biologically significant in sperm [85] [83].
- Sensitivity for Low Input: Compatible with DNA yields from challenging sample types like fine needle aspirates and FFPE tissues, suggesting utility for sperm samples where DNA quantity may be limited [84].
- Contamination Assessment: Can be targeted to specific biomarker CpGs to quantify the level of somatic cell contamination in sperm samples, a major confounder in sperm epigenetics [2].
Detailed Experimental Protocol
Step 1: Bisulfite Conversion
- Extract genomic DNA from sperm samples, employing methods that include somatic cell lysis or post-extraction quality checks to minimize somatic DNA contamination [2].
- Convert 500 ng to 1 µg of DNA using a commercial bisulfite conversion kit (e.g., EZ DNA Methylation Kit, Zymo Research). Validate conversion efficiency by ensuring >99% conversion of non-CpG cytosines.
Step 2: PCR Amplification
- Design PCR primers that flank the CpG site(s) of interest identified from the Infinium BeadChip analysis. Primer design considerations are critical [83]:
- Amplicon Length: 80-200 bp to minimize DNA fragmentation from bisulfite conversion.
- Primer Specificity: Each primer should contain at least four non-CpG cytosines to ensure binding only to bisulfite-converted DNA.
- Avoid CpGs: Primers should not contain CpG dinucleotides to prevent biased amplification. If unavoidable, use degenerate bases.
- Biotin Labeling: One primer must be 5'-end labeled with biotin for subsequent bead immobilization. HPLC purification is recommended.
- Perform PCR amplification under standard conditions, optimizing cycles to avoid plateau phase.
Step 3: Pyrosequencing
- Bind the biotinylated PCR product to streptavidin-coated sepharose beads.
- Wash and denature the double-stranded DNA to obtain a single-stranded template.
- Hybridize a sequencing primer to the template, upstream of the CpG site(s) to be analyzed.
- Load the prepared template into a pyrosequencer (e.g., Pyromark Q48 or Q96, Qiagen).
- The instrument sequentially dispenses nucleotides in a predefined order. The incorporation of a complementary nucleotide releases pyrophosphate (PPi), which is converted to a light signal via a series of enzymatic reactions. The light intensity is proportional to the number of nucleotides incorporated [83] [84].
- Analyze the resulting pyrogram. The methylation percentage at each CpG is calculated as:
% Methylation = [C Peak Height / (C Peak Height + T Peak Height)] * 100[83].
Technical Validation and Quality Control
For rigorous assay validation, the following performance figures should be established, especially when quantifying low methylation levels or subtle differences [85]:
- Limit of Blank (LoB): The highest apparent methylation measured in a blank sample.
- Limit of Detection (LoD): The lowest methylation level that can be reliably distinguished from the LoB.
- Limit of Quantification (LoQ): The lowest methylation level that can be quantified with acceptable precision and accuracy.
- Precision: Assessed via repeatability (intra-run) and reproducibility (inter-run) measurements [84].
Table 1: Key Performance Characteristics of Pyrosequencing
| Characteristic | Description | Typical Performance/Value |
|---|---|---|
| Resolution | Single-base resolution for CpG sites within an amplicon | Quantitative data for each CpG in a sequenced region [83] |
| Throughput | Number of samples and loci processed per run | Medium; ideal for validating tens to hundreds of loci across many samples [83] |
| Accuracy | Concordance with known methylation standards | High; often used as a reference method [83] |
| Precision | Reproducibility of measurements | High; CVs typically <5-10% [85] |
| DNA Input | Amount of DNA required post-bisulfite conversion | 10-50 ng per PCR reaction [83] |
Diagram 1: Pyrosequencing Workflow for DNA Methylation Analysis. The process begins with DNA extraction and bisulfite conversion, followed by PCR with a biotinylated primer, template preparation, and sequential nucleotide incorporation with real-time light detection for quantification.
CHARM for Genome-Wide Methylation Validation
Comprehensive High-Throughput Arrays for Relative Methylation (CHARM) is a microarray-based method for epigenome-wide methylation analysis. Unlike the Infinium BeadChip, CHARM is not restricted to pre-defined CpG sites and uses methylation-dependent fractionation via the McrBC enzyme, followed by hybridization to a custom tiling array [86] [87]. McrBC cleaves DNA at methylated cytosine residues (recognition site RmC(N)~55-103-RmC), thereby fractionating the genome into methylated (cleaved) and unmethylated (intact) portions. The intact, unmethylated DNA is then competitively hybridized to an array, allowing for the identification of differentially methylated regions (DMRs) without a priori assumptions about their location, making it excellent for validating and discovering novel DMRs outside of traditional CpG islands [86] [87].
Advantages for Sperm Epigenetics
- CpG Density Independence: Effectively measures methylation in CpG-poor regions, such as CpG "shores" and "shelves," which are often informative and may be under-represented on standard arrays [86] [87].
- Discovery-Oriented: As a validation tool, it can confirm DMRs found by BeadChip and identify additional relevant regions that the array may have missed due to probe design limitations.
- Quantitative for Regions: Provides quantitative data across genomic regions rather than at single CpGs, offering a complementary perspective to pyrosequencing.
Detailed Experimental Protocol
Step 1: DNA Fractionation with McrBC
- Shear 3.5–7 µg of genomic DNA (e.g., using a sonicator) to a random fragment distribution.
- Digest the sheared DNA with McrBC enzyme. McrBC will cleave DNA containing methylated cytosines, while unmethylated DNA remains intact.
- Separate the digested DNA by gel electrophoresis. The high molecular weight (unmethylated) fraction is gel-purified. This is the "methyl-depleted" (MD) fraction. The "untreated" (UT) fraction is a portion of the sheared DNA that did not undergo McrBC digestion [87].
Step 2: DNA Labeling and Hybridization
- Label the UT DNA fraction with Cyanine-3 (Cy3) and the MD fraction with Cyanine-5 (Cy5).
- Co-hybridize the labeled UT and MD samples to a CHARM microarray (e.g., NimbleGen HD2 platform). The array is designed with tiling probes across genomic regions of interest, including non-CpG island regions [86] [87].
- Wash the array to remove non-specifically bound DNA.
Step 3: Data Acquisition and Analysis
- Scan the array to obtain fluorescence intensities for Cy3 (UT, total DNA) and Cy5 (MD, unmethylated DNA) for each probe.
- The primary measurement is the log2 ratio (M-value) of Cy3 to Cy5 intensities, which reflects the relative methylation level [87].
- Normalization: Use a modified loess normalization based on control probes from CpG-free regions that are guaranteed to be unmethylated and uncut by McrBC.
- Smoothing: Apply a moving window smoother to reduce noise and probe-effect biases, generating a smoothed M-value for each genomic location.
- DMR Calling: Identify statistically significant Differentially Methylated Regions (DMRs) between sample groups (e.g., infertile vs. fertile sperm) using Z-scores and false discovery rates (FDR) derived from permutation testing [87].
Table 2: Comparison of Methylation Assessment Techniques
| Parameter | Infinium MethylationEPIC v2 | Pyrosequencing | CHARM |
|---|---|---|---|
| Principle | BeadChip hybridization after bisulfite conversion | Sequencing-by-synthesis of bisulfite-converted DNA | Array hybridization after methylation-dependent fractionation |
| Resolution | Single CpG (pre-designed) | Single CpG (within amplicon) | Regional (100s of bp) |
| Genome Coverage | ~935,000 pre-selected CpG sites [4] | User-defined targets | Genome-wide, agnostic to CpG density [86] [87] |
| Quantitation | Beta-value (0-1) | Percentage (0-100%) | Log2 ratio (M-value) |
| Best Use | Primary discovery | Targeted, high-precision validation | Genome-wide validation & discovery in non-CpG island regions |
Diagram 2: CHARM Array Workflow. The procedure involves fragmenting genomic DNA, digesting with the methylation-sensitive McrBC enzyme, purifying the unmethylated fraction, and performing two-color competitive hybridization on a tiling array to identify regions of differential methylation.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Methylation Validation
| Reagent / Kit | Function | Application Notes |
|---|---|---|
| EZ DNA Methylation Kit (Zymo Research) | Bisulfite conversion of genomic DNA. | Critical for pyrosequencing; converts unmethylated C to U while leaving 5mC intact [48] [83]. |
| PyroMark PCR Kit (Qiagen) | PCR amplification of bisulfite-converted DNA. | Optimized for bisulfite templates; includes biotinylated primers for pyrosequencing [83] [84]. |
| PyroMark Q48 Autoprep System (Qiagen) | Integrated instrument and reagents for pyrosequencing. | Compact platform for automated template preparation and sequencing; suitable for clinical samples [84]. |
| McrBC Enzyme (NEB) | Methylation-dependent restriction enzyme. | Core of CHARM fractionation; cleaves DNA containing methylated cytosines [86] [87]. |
| CHARM HD2 Microarray (Roche NimbleGen) | Custom tiling array for hybridization. | Provides broad, unbiased coverage of the genome, including non-promoter regions [87]. |
| NimbleGen Labeling Kit (Roche NimbleGen) | Fluorescent dye labeling of DNA. | For labeling UT and MD fractions with Cy3 and Cy5 for CHARM array hybridization [87]. |
| Somatic Cell Lysis Buffer | Selective lysis of somatic cells in semen. | Crucial pre-analytical step in sperm epigenetics to minimize confounding methylation signals from somatic DNA [2]. |
The integration of orthogonal validation methods is a non-negotiable component of robust sperm epigenetics research utilizing the Infinium Methylation BeadChip. Pyrosequencing stands out for its unparalleled quantitative accuracy in confirming methylation levels at specific CpG sites of high interest, while CHARM offers a powerful approach for validating and extending discoveries across the methylome in an unbiased manner. Employing these techniques in tandem, with careful attention to sperm-specific challenges like somatic cell contamination, ensures that reported methylation signatures are reliable and biologically meaningful, thereby strengthening conclusions related to male infertility and environmental impacts on the sperm epigenome.
Conclusion
The Infinium Methylation BeadChip stands as a powerful, cost-effective tool for profiling the sperm methylome, with strong evidence linking paternal epigenetic marks to offspring health. The latest EPICv2 array offers significant refinements, including better probe mapping and support for diverse populations. However, researchers must remain vigilant about technical noise and probe reliability. Future directions should focus on developing sperm-specific bioinformatic tools, expanding longitudinal studies to solidify causal links, and translating these epigenetic discoveries into clinical applications for predictive diagnostics and understanding intergenerational disease risk.
References
- 1. High-resolution analyses of human sperm dynamic ... [https://clinicalepigeneticsjournal.biomedcentral.com/articles/10.1186/s13148-020-00988-1]
- 2. Tackling somatic DNA contamination in sperm epigenetic ... [https://pubmed.ncbi.nlm.nih.gov/39974749/]
- 3. The combined effect of obesity and aging on human sperm ... [https://www.nature.com/articles/s41598-020-71979-8]
- 4. Comprehensive evaluation of the Infinium human ... [https://epicom.biomedcentral.com/articles/10.1186/s43682-023-00021-5]
- 5. Ewastools: Infinium Human Methylation BeadChip pipeline ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7219210/]
- 6. Infinium Mouse Methylation BeadChip Kit [https://www.illumina.com/products/by-type/microarray-kits/infinium-mouse-methylation.html]
- 7. DNA methylation-based semen age prediction using the ... [https://www.sciencedirect.com/science/article/abs/pii/S1872497325000237]
- 8. Epigenetic age prediction in semen – marker selection and ... [https://www.aging-us.com/article/203399/text]
- 9. Methylation Array Data Analysis Tips [https://www.illumina.com/techniques/microarrays/methylation-arrays/methylation-array-data-analysis-tips.html]
- 10. Histology, Spermatogenesis - StatPearls - NCBI Bookshelf - NIH [https://www.ncbi.nlm.nih.gov/books/NBK553142/]
- 11. Spermatogenesis [https://en.wikipedia.org/wiki/Spermatogenesis]
- 12. Spermatogenesis - Developmental Biology - NCBI Bookshelf [https://www.ncbi.nlm.nih.gov/books/NBK10095/]
- 13. Sperm is epigenetically programmed to regulate gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4971762/]
- 14. The role of genomic imprinting in biology and disease [https://www.nature.com/articles/nrg3766]
- 15. The role of genomic imprinting in biology and disease [https://pubmed.ncbi.nlm.nih.gov/24958438/]
- 16. Determining environmental causes of biological effects [https://pmc.ncbi.nlm.nih.gov/articles/PMC3350663/]
- 17. Epigenetic changes in sperm are associated with paternal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12450096/]
- 18. Epigenetic regulation by DNA methylation, histone ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12500515/]
- 19. Genetic susceptibility to neurodevelopmental conditions ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11245083/]
- 20. Tackling somatic DNA contamination in sperm epigenetic ... [https://www.frontiersin.org/journals/reproductive-health/articles/10.3389/frph.2025.1506117/full]
- 21. An epigenome-wide association study in the case-control ... [https://jneurodevdisorders.biomedcentral.com/articles/10.1186/s11689-025-09637-1]
- 22. Evaluation of the pooled sample method in Infinium ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10463626/]
- 23. Emerging evidence that the mammalian sperm epigenome ... [https://www.nature.com/articles/s41467-023-37820-2]
- 24. Characterisation and reproducibility of the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10916044/]
- 25. Infinium MethylationEPIC v2.0 Kit | Methylation profiling array [https://www.illumina.com/products/by-type/microarray-kits/infinium-methylation-epic.html]
- 26. High-resolution analyses of human sperm dynamic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7735420/]
- 27. Age-related methylation changes in the human sperm ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10042684/]
- 28. Sperm Ribosomal DNA Promoter Methylation Levels Are ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2020.00319/full]
- 29. Age-related methylation changes in the human sperm ... [https://www.aging-us.com/article/204546/text]
- 30. Technical variability across the 450K, EPICv1, and EPICv2 ... [https://clinicalepigeneticsjournal.biomedcentral.com/articles/10.1186/s13148-024-01761-4]
- 31. DNAm age differences between infinium ... [https://www.nature.com/articles/s42003-025-08021-y]
- 32. Sperm DNA methylation mediates the association of male ... [https://www.nature.com/articles/s41598-020-80857-2]
- 33. Functional assessment and comparison of different ... [https://www.sciencedirect.com/science/article/abs/pii/S0921448825001385]
- 34. Sperm Separation and Selection Techniques to Mitigate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11856810/]
- 35. Semen Collection and Analysis [https://bio-protocol.org/exchange/minidetail?id=10426786&type=30]
- 36. Comparison of current methods for genome-wide DNA ... [https://epigeneticsandchromatin.biomedcentral.com/articles/10.1186/s13072-025-00616-3]
- 37. Epigenetic age prediction in semen – marker selection and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8386575/]
- 38. Comparative performance evaluation of bisulfite [https://pmc.ncbi.nlm.nih.gov/articles/PMC11969950/]
- 39. Ultra-mild bisulfite outperforms existing methods for 5- ... [https://www.nature.com/articles/s41467-025-66033-y]
- 40. Bisulfite-Converted DNA Quantity Evaluation: A Multiplex ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7947210/]
- 41. Comparison of enzymatic and bisulfite conversion of ... [https://clinicalepigeneticsjournal.biomedcentral.com/articles/10.1186/s13148-025-01901-4]
- 42. Abnormal methylation of ST8SIA4 in low-motility sperm [https://www.sciencedirect.com/science/article/pii/S1028455925001305]
- 43. Analysis of DNA Methylation by Bisulfite Conversion [https://www.neb.com/en-us/products/epigenetics/methylation-analysis/bisulfite-conversion?srsltid=AfmBOooY5HNF5bbQ_buUe5rBMh0Cp3t5oJaCOVwT8hwAjFnGh28GOFDp]
- 44. DNA Methylation Profiling Using Infinium Methylation Assay [https://bio-protocol.org/en/bpdetail?id=777&type=0]
- 45. Introduction [https://support-docs.illumina.com/ARR/Inf-MSA/Content/ARR/MSA/Intro.htm]
- 46. Characterisation and reproducibility of the ... - BMC Genomics [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10027-5]
- 47. Infinium MethylationEPIC BeadChip Documentation [https://support.illumina.com/array/array_kits/infinium-methylationepic-beadchip-kit/documentation.html]
- 48. Comparison of current methods for genome-wide DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12376410/]
- 49. Comprehensive analysis of DNA methylation patterns in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12513379/]
- 50. Analysis of 450k data using minfi [https://www.bioconductor.org/help/course-materials/2015/BioC2015/methylation450k.html]
- 51. Comparison of pre-processing methodologies for Illumina ... [https://clinicalepigeneticsjournal.biomedcentral.com/articles/10.1186/s13148-016-0241-2]
- 52. SeSAMe: reducing artifactual detection of DNA methylation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6237738/]
- 53. Comparative epigenome analysis using Infinium DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10147478/]
- 54. An effective processing pipeline for harmonizing DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8424820/]
- 55. Accounting for differences between Infinium ... [https://www.life-science-alliance.org/content/8/9/e202403155]
- 56. New measure of sperm age may be predictor of pregnancy ... [https://today.wayne.edu/medicine/news/2022/05/13/new-measure-of-sperm-age-may-be-predictor-of-pregnancy-success-47890]
- 57. Environmental factors accelerate epigenetic aging of ... [https://www.sciencedirect.com/science/article/pii/S2666335X25000680]
- 58. Type 2 diabetes impacts DNA methylation in human sperm | Clinical Epigenetics | Full Text [https://clinicalepigeneticsjournal.biomedcentral.com/articles/10.1186/s13148-025-01853-9]
- 59. SURF: A Self‐Supervised Deep Learning Method for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12631812/]
- 60. Associations between Sperm Epigenetic Age and Semen ... [https://www.mdpi.com/1467-3045/46/2/101]
- 61. Troubleshooting [https://support-docs.illumina.com/ARR/InfLabSetup/Content/ARR/SetupandProcedures/fINF_Troubleshooting.htm]
- 62. Hybridization and X stain Infinium Assay Troubleshooting ... [https://knowledge.illumina.com/microarray/general/microarray-general-troubleshooting-list/000005025]
- 63. Low-input and single-cell methods for Infinium DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11040145/]
- 64. Technical and biological sources of unreliability of Infinium ... [https://clinicalepigeneticsjournal.biomedcentral.com/articles/10.1186/s13148-024-01739-2]
- 65. The impact of low input DNA on the reliability ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9665153/]
- 66. How to Optimize Data Analysis from Methylation Arrays [https://www.cd-genomics.com/resource-methylation-arrays-data-analysis-tips.html]
- 67. Epigenetics of Male Infertility: The Role of DNA Methylation [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2021.689624/full]
- 68. Cross-reactive probes on Illumina DNA methylation arrays [https://pmc.ncbi.nlm.nih.gov/articles/PMC7745769/]
- 69. Evaluation of the HumanMethylationEPIC v2.0 Bead Chip ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12369268/]
- 70. Creation and validation of the first infinium DNA methylation ... [https://epicom.biomedcentral.com/articles/10.1186/s43682-024-00028-6]
- 71. Batch-effect detection, correction and characterisation in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9055778/]
- 72. Annotation for Illumina's EPIC v2.0 methylation arrays [https://jokergoo.github.io/IlluminaHumanMethylationEPICv2anno.20a1.hg38/]
- 73. Reproducibility and Reliability of Semen Analyses in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3712861/]
- 74. DNA Methylation Validation Methods: a Coherent Review ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6771119/]
- 75. How to Validate DNA Methylation Sequencing Results [https://www.cd-genomics.com/resource-how-to-validate-dna-methylation-sequencing-results.html]
- 76. Accounting for differences between Infinium ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12238762/]
- 77. Accounting for differences between Infinium ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11245009/]
- 78. Sperm epigenetics and influence of environmental factors - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6034033/]
- 79. MethylCallR : a comprehensive analysis framework for ... [https://www.nature.com/articles/s41598-024-77914-5]
- 80. Comparative Performance of Methylation Array and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12589796/]
- 81. Cost-effective solutions for high-throughput enzymatic DNA ... [https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1011667]
- 82. Cost-effective promoter methylation analysis via target long ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12309061/]
- 83. DNA Methylation Validation Methods: a Coherent Review with ... [https://biologicalproceduresonline.biomedcentral.com/articles/10.1186/s12575-019-0107-z]
- 84. Validation of a Compact and Self-Contained ... [https://www.mdpi.com/2075-4418/15/3/390]
- 85. Quantitative Validation and Quality Control of ... [https://pubmed.ncbi.nlm.nih.gov/26103890/]
- 86. Comprehensive high-throughput arrays for relative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2336799/]
- 87. Comprehensive High-Throughput Arrays for Relative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6353553/]